ANEMIA
↓ in RBC mass: Hct <41% or Hb <13.5 g/dL (men); Hct <36% or Hb <12 g/dL (women)
Clinical manifestations
• Symptoms: ↓ O2 delivery → fatigue, exertional dyspnea, angina (if CAD)
• Signs: pallor (mucous membranes, palmar creases), tachycardia, orthostatic hypotension
• Other findings: jaundice (hemolysis), splenomegaly (thalassemia, neoplasm, chronic hemolysis), petechiae/purpura (bleeding disorder), glossitis (iron, folate, vitamin B12 defic.), koilonychia (iron defic.), neurologic abnormalities (B12 defic.)
Diagnostic evaluation
• History: bleeding, systemic illness, drugs, exposures, alcohol, diet (including pica), FHx
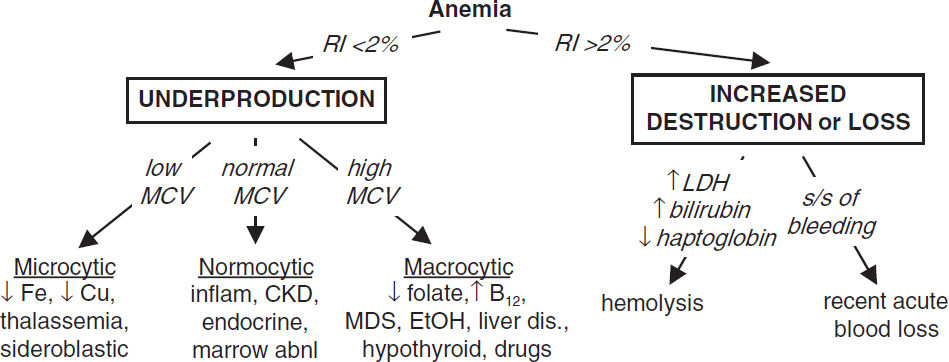
• CBC w/ diff.; RBC params incl. retics, MCV (nb, mixed disorder can → nl MCV), RDW
• Reticulocyte index (RI) = [reticulocyte count × (Pt’s Hct/nl Hct)]/maturation factor maturation factors for a given Hct: 45% = 1, 35% = 1.5, 25% = 2, 20% = 2.5 RI >2% → adequate marrow response; RI <2% → hypoproliferation
• Peripheral smear: select area where roughly ⅓ RBCs touch each other; ✓ RBC size, shape, inclusions (see “Appendix” & “Peripheral Smear”), WBC morphology, plt count
• Additional labs as indicated: hemolysis labs (if RI >2%, see below), iron/TIBC, ferritin, folate, B12, LFTs, BUN, & Cr, TFTs, Hb electrophoresis, enzyme/gene mutation screens
• Bone marrow (BM) aspirate and biopsy (bx) with cytogenetics as indicated
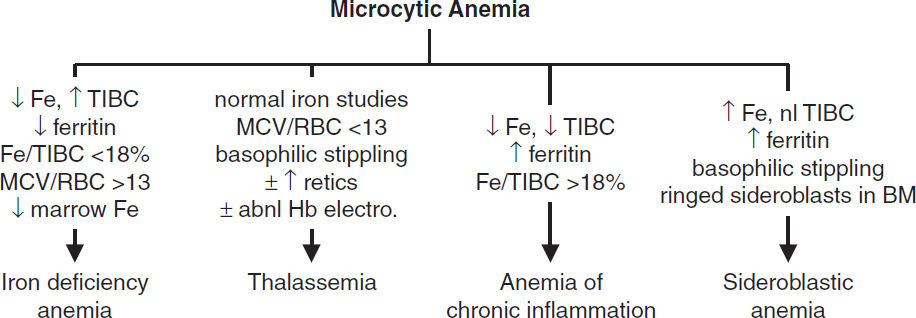
MICROCYTIC ANEMIAS
Iron deficiency (Lancet 2021;397:233)
• ↓ marrow iron & depleted body iron stores → ↓ heme synthesis → microcytosis → anemia
• Special clinical manifestations: angular cheilosis, atrophic glossitis, pica (consumption of nonnutritive substances such as ice, clay), koilonychia (nail spooning)
Plummer-Vinson syndrome (iron deficiency anemia, esophageal web & atrophic glossitis)
• Etiologies: chronic bleeding (GI—incl. cancer, menstrual, parasites, NSAIDs, etc.), ↓ supply (malnutrition; ↓ absorp. due to celiac sprue, Crohn’s, ↑ gastric pH, subtotal gastrectomy), ↑ demand (preg; Blood 2017;129:940). Iron-refractory iron-defic. anemia (IRIDA; rare genetic disorder due to hepcidin dysregulation; Nat Genet 2008;40:569).
• Diagnosis (eval ideally before Rx): ↓ Fe, ↑ TIBC, ↓ ferritin (esp. <15), ↓ transferrin sat (Fe/TIBC; esp. <15%), ↑ soluble transferrin receptor; ↑ plt. Unless hx c/w other etiology, initiate workup for GIB, incl. H. pylori serology. ? Celiac labs (anti-TTG, anti-endomysial IgA Abs). Cytogenetics & molecular testing as indicated (eg, MDS, leukemia).
• Treatment: oral Fe TID (~6 wks to correct anemia; ~6 mo to replete Fe stores; nb, oral Fe does not give ⊕ Hemoccult). With severe anemia, persistent losses, prior to Epo Rx, or while inpatient, use IV iron (Fe-sucrose, -gluconate, -dextran).
Thalassemias (Lancet 2018;391:155)
• ↓ synthesis of α- or β-globin chains of Hb → ≠ subunits → destruction of RBCs and erythroid precursors; ∴ anemia from hemolysis and ineffective erythropoiesis
• α-thalassemia (NEJM 2014;371:1908): deletions in α-globin gene complex (nl 4 α genes), seen w/ Southeast Asian, Mediterranean, African, Middle East ancestry
3 α → silent carrier; 2 α → α-thal minor = mild anemia, α-thal-1 (--/αα, milder) or
α-thal-2 (-α/-α); 1 α → HbH (β4) disease = severe anemia, hemolysis, and splenomegaly
0 α genes → Hb Barts (γ4) = intrauterine hypoxia and hydrops fetalis
• β-thalassemia: mutations in β-globin gene → absent or ↓ gene product seen w/ Mediterranean (espec. Greek or Italian), African, or Asian ancestry
1 mutated β gene → thal minor (or trait) = mild anemia (no transfusions)
2 mutated β genes → thal intermedia (occasional transfusions) or thal major (= Cooley’s anemia; transfusion dependent) depending on severity of mutations
• Special clinical manifestations: chipmunk facies, pathologic fractures, hepatosplenomegaly (due to extramedullary hematopoiesis), high-output CHF, bilirubin gallstones, Fe overload
• Dx: MCV <70, normal Fe, ferritin, MCV/RBC count <13 [Mentzer Index, 60% Se, 98% Sp; (Ann Hem 2007;86:486)], ± ↑ retics, basophilic stippling; Hb electrophoresis: ↑ HbA2 (α2δ2) in β-thal; normal pattern in α-thal trait, ∴ PCR or supravital stain for dx
• Treatment: folate; transfusions + Fe chelator [either deferoxamine (IV) or deferasirox (PO)]; ? splenectomy if ≥50% ↑ in transfusions; gene therapy in development (NEJM 2018;378:1479); luspatercept (↓ SMAD signaling) in β-thal major (NEJM 2020;382:1219)
Anemia of chronic inflammation (see below)
Sideroblastic anemia
• Defective heme biosynthesis within RBC precursors
• Etiologies: hereditary/X-linked (ALAS2 mutations), idiopathic, MDS-RARS, reversible (alcohol, lead, isoniazid, chloramphenicol, copper deficiency, hypothermia)
• Special clinical manifestations: hepatosplenomegaly, iron overload syndromes
• Dx: social, work, & TB hx; can be micro-, normo-, or macrocytic; variable populations of hypochromic RBCs; ↑ Fe, nl TIBC, ↑ ferritin, basophilic stippling, RBC Pappenheimer bodies (Fe-containing inclusions), ring sideroblasts (w/ iron-laden mitochondria) in BM
• Treatment: treat reversible causes; trial of pyridoxine, supportive transfusions for severe anemia with chelation therapy; high-dose pyridoxine for some hereditary cases
NORMOCYTIC ANEMIAS
Anemia of chronic inflammation (ACI; NEJM 2012;366:4)
• ↓ RBC production due to impaired iron utilization and functional iron deficiency from ↑ hepcidin; cytokines (IL-6, TNF-α) cause ↓ Epo responsiveness/production
• Etiologies: autoimmune disorders, chronic infection, inflammation, HIV, malignancy
• Dx: ↓ Fe, ↓ TIBC (usually normal or low transferrin sat), ± ↑ ferritin; usually normochromic, normocytic (~70% of cases) but can be microcytic if prolonged
• Coexisting iron deficiency common. Dx clues include ↓ serum ferritin levels, absence of iron staining on BM bx, ⊕ response to a trial of oral iron and/or ↑ soluble transferrin receptor/ferritin index (Am J Clin Pathol 2012;138:642).
• Treatment: treat underlying disease ± iron and/or erythropoiesis-stimulating agent (ESA; eg, Epo). Iron if ferritin <100 or Fe/TIBC <20%. Consider ESA if Epo <500. Avoid ESA in cancer if treatment goal is cure (Lancet 2009;373:1532). Transfuse PRBCs only if symptomatic & insufficient time to wait for response to Epo or underlying disease Rx.
Anemias of other chronic disorders
• Anemia of CKD: ↓ Epo; treat w/ Epo & iron (see “Chronic Kidney Disease”)
• Endocrine deficiencies: hypometabolism and ↓ O2 demand with thyroid, pituitary, adrenal, or parathyroid disease → ↓ Epo; can be normocytic or macrocytic
Sideroblastic anemia (see above)
Pure red cell aplasia
• Destructive antibodies or lymphocytes → ineffective erythropoiesis
• Associated with thymoma, CLL, parvovirus infection, autoimmunity, drugs
• Diagnostic studies: lack of erythroid precursors on BM bx, other lines normal
• Treatment: thymectomy if thymus enlarged; IVIg if parvovirus and immunosuppressed (Clin Infect Dis 2013;56:968); immunosuppression/chemoRx if CLL or idiopathic; supportive care w/ PRBC transfusions; ? erythropoietin receptor agonist if due to antierythropoietin Ab (NEJM 2009;361:1848); consider hematopoietic cell transplantation.
includes megaloblastic and nonmegaloblastic causes
Megaloblastic anemia
• Impaired DNA synthesis → cytoplasm matures faster than nucleus → ineffective erythropoiesis and macrocytosis; due to folate or B12 deficiency; also in MDS
• ✓ folate and vitamin B12; ↑ LDH & indirect bilirubin (due to ineffective erythropoiesis)
• Smear: neutrophil hypersegmentation, macro-ovalocytes, anisocytosis, poikilocytosis
Folate deficiency
• Folate present in leafy green vegetables and fruit; total body stores sufficient for 2–3 mo
• Etiologies: malnutrition (alcoholics, anorectics, elderly), ↓ absorption (sprue), impaired metabolism (methotrexate, pyrimethamine, trimethoprim; NEJM 2015;373:1649), ↑ requirement (chronic hemolytic anemia, pregnancy, malignancy, dialysis)
• Diagnosis: ↓ folate; ↓ RBC folate, ↑ homocyst. but nl methylmalonic acid (unlike B12 defic.)
• Treatment: folate 1–5 mg PO qd for 1–4 mo or until complete hematologic recovery; critical to r/o B12 deficiency first (see below)
Vitamin B12 deficiency (NEJM 2013;368:149)
• B12 present only in foods of animal origin; total body stores sufficient for 2–3 y
• Binds to intrinsic factor (IF) secreted by gastric parietal cells; absorbed in terminal ileum
• Etiologies: malnutrition (alcoholics, vegans), pernicious anemia (PA, autoimmune disease against gastric parietal cells, a/w polyglandular endocrine insufficiency and ↑ risk of gastric carcinoma), other causes of ↓ absorption (gastrectomy, sprue, Crohn’s disease), ↑ competition (intestinal bacterial overgrowth, fish tapeworm)
• Clinical manifestations: neurologic changes (subacute combined degeneration) affecting peripheral nerves, posterior & lateral columns of the spinal cord and cortex → numbness, paresthesias, ↓ vibratory & positional sense, ataxia, dementia, mood
• Dx: ↓ B12 (even low nl); ↑ homocysteine & methylmalonic acid; anti-IF Ab; ↑ gastrin in PA
• Treatment: 1 mg B12 IM qd × 7 d → q wk × 4–8 wk → q month for life
neurologic abnormalities are reversible if treated w/in 6 mos
folate can reverse hematologic abnormalities of B12 deficiency but not neurologic changes (and can “steal” B12 stores → worsening of neuro complications)
oral supplementation (2 mg qd) appears feasible as well (Cochrane Rev CD004655) even w/o IF
Nonmegaloblastic macrocytic anemias
• Liver disease: often macrocytic, may see target cells, or spur cell anemia w/ hemolysis
• Alcoholism: BM suppression & macrocytosis independent of folate/B12 defic. or cirrhosis
• Other causes: reticulocytosis; hypothyroid; MDS; meds impairing DNA synth (zidovudine, 5-FU, hydroxyurea, Ara-C); hereditary orotic aciduria; Lesch-Nyhan syndrome
PANCYTOPENIA
Etiologies
• Hypocellular bone marrow (nl cellularity ~100 – age): aplastic anemia, hypoplastic MDS
• Cellular bone marrow: MDS, aleukemic leukemia, PNH, severe megaloblastic anemia
• Myelophthesis (marrow replacement, PMF); systemic dis. (hypersplen, sepsis, EtOH/toxin)
Clinical manifestations
• Anemia → fatigue; neutropenia → recurrent infections; thrombocytopenia → mucosal bleeding & easy bruisability
Aplastic anemia (stem cell failure) (NEJM 2015;373:35)
• Epidemiology: 2–5 cases/106/y; biphasic (major peak in adolescents, 2nd peak in elderly)
• Diagnosis: pancytopenia w/ ↓ retics, BM bx w/ hypocellularity, usually nl cytogenetics
• Etiologies: idiopathic (½ – ⅔ of cases)
Stem cell destruction: radiation, chemotherapy, chemicals (eg, benzene)
Med rxn (eg, chloramphenicol, NSAIDs, sulfa drugs, gold, carbamazepine, antithyroid)
Viruses (HHV-6, HIV, EBV, parvovirus B19); post-viral hepatic failure (not Hep A/B/C)
Immune disorders (SLE, GVHD post-HSCT, thymoma)
PNH (see below); Fanconi’s anemia (congenital disorder w/ pancytopenia, macrocytic anemia, ↑ risk of MDS, AML, & SCC of head & neck, and multiple physical anomalies)
Shortened telomeres: seen w/ telomerase (TERT, TERC) mut. (10% of aplastic anemia), dyskeratosis congenita/DKC1 mut; a/w IPF, cirrhosis (NEJM 2009;361:2353)
Somatic mutations: PNH clones in ~50% of aplastic anemia (Haematologica 2010;95:1075)
• Treatment and prognosis
Immunosuppression (CsA/tacrolimus, ATG): 70–80% respond, with 80–90% 5-y survival in responders (96% vs. 76% w/ horse vs. rabbit ATG; NEJM 2011;365:430); 15–20% 10-y incidence of clonal disorders (mostly MDS, AML, PNH)
TPO mimetics (eg, eltrombopag): use 1st-line w/ immunosuppression (NEJM 2022;386:11)
Supportive care: transfusions, abx, possible utility of G-CSF & Epo (if Epo <500)
Allogeneic HSCT: for young Pts → ~80% long-term survival and significantly ↓ risk of malignant evolution, but has risk of transplant-related morbidity & mortality; if possible, avoid transfusions (risk of alloimmunization) pretransplant
Myelodysplastic syndromes (MDS) (qv)
Paroxysmal nocturnal hemoglobinuria (PNH) (Blood 2009;113:6522)
• Acquired clonal stem cell disorder = inactivating somatic mutation of PIG-A gene → deficiency of GPI-anchor for CD55 & CD59 (inhib of complement) → complement-mediated RBC lysis, plt aggregation, & hypercoagulability
• Clinical: intravascular hemolytic anemia, hypercoagulability (venous >arterial; esp. intraabdominal, cerebral), smooth muscle dystonias, deficient hematopoiesis (cytopenias); a/w aplastic anemia, MDS and evolution to AML
• Dx: flow cytometry (↓ CD55 & CD59) on RBCs and granulocytes; urine hemosiderosis
• Treatment: supportive care (iron, folate, transfusions); consider anticoagulation.
Allogeneic HSCT for hypoplasia or severe thrombosis.
Pegcetacoplan (binds C3, prevents complement cascade activation) superior to eculizumab (Ab inactivates terminal complement C5s) in ↓ hemolysis & stabilizing Hb levels (NEJM 2021;384:1028). Eculizumab effective in pregnancy (NEJM 2015;373:1032); must have meningococcal vaccination.
Myelophthisic anemia (see also “Primary Myelofibrosis”)
• Infiltration of bone marrow by cancer (commonly metastatic solid tumors), leukemia, infection, fibrosis (primary myelofibrosis), granulomas, lysosomal storage disorders
HEMOLYTIC ANEMIAS
Causes of Hemolytic Anemia by Mechanism (Lancet 2000;355:1169 & 1260) |
|||
Cause |
Mechanism |
Examples |
Mode |
Intrinsic |
Enzyme deficiency |
G6PD deficiency |
Hereditary |
Hemoglobinopathies |
Sickle cell anemia, thalassemia |
||
Membrane abnormalities |
Hereditary spherocytosis |
||
PNH, spur cell anemia in liver disease |
Acquired |
||
Extrinsic |
Immune-mediated |
Autoimmune; drug-induced, tx rxn |
|
Traumatic |
MAHA; prostheses (valves, TIPS) |
||
Direct infections, toxins |
Malaria, babesiosis; snake & spider venoms; Wilson’s; hypotonic infusions |
||
Entrapment |
Hypersplenism |
||
Diagnostic evaluation
• ↑ retic count (RI >2%), ↑ LDH, ↓ hapto (83% Se, 96% Sp), ↑ indirect bili, ✓ vit C & Cu
• Autoimmune hemolysis: Coombs’ test = direct antiglobulin test (DAT) → ⊕ if agglutination occurs when antisera against Ig or C3 are applied to patient RBCs
• Location of hemolysis (many conditions can include components of both)
Intravascular: RBC destruction in vessels (shear by mech valve, DIC, toxins); assoc. w/ hemoglobinemia, hemoglobinuria, hemosiderinuria, ↑↑ LDH, ↓ haptoglobin.
Extravascular: more common cause. Mϕ clear damaged/opsonized RBC; splenomegaly (reticuloendothelial expansion in spleen, liver, BM, LNs); ↑ LDH ↓ hapto
• Family h/o anemia; personal or family h/o cholelithiasis
Glucose-6-phosphate dehydrogenase (G6PD) deficiency (Lancet 2008;371:64)
• X-linked defect of metabolism (G6PD mutations) w/ ↑ susceptibility to oxidative damage
• Most common in ♂ of African or Mediterranean descent (malaria-endemic areas)
• Hemolysis precipitated by drugs (sulfonamides, dapsone, nitrofurantoin, rasburicase, primaquine, doxorubicin, methylene blue), infxn, DKA, foods (favism, NEJM 2018;378:60)
• Diagnosis: smear may show RBC Heinz bodies (oxidized Hb) that result in bite cells once removed by spleen; ↓ G6PD levels (may be normal after acute hemolysis because older RBCs have already lysed and young RBCs may still have near-normal levels)
Sickle cell anemia (NEJM 2017;376:1561 & Lancet 2017;390:311)
• Recessive β-globin mutation → structurally abnl hemoglobin (HbS). ~8% African Americans heterozygotes (“sickle trait”; usually w/o sx); ~1/400 homozygotes (sickle cell disease).
• ↓ O2 → HbS polymerizes → RBC sickles, ↓ RBC deformability → hemolysis & microvascular occlusion due to endothelial activ. & PMN adhesion (Blood 2013;122:3892)
• Anemia: chronic hemolysis ± acute aplastic (parvo. B19) or splenic sequestration crises
• Vaso-occlusion & infarction: acute chest syndrome & stroke (high mortality), pulmonary HTN, painful crises, splenic sequestration, renal papillary necrosis, avascular necrosis, dactylitis (hand–foot syndrome), priapism
• Infection: splenic infarction → overwhelming infection by encapsulated organisms; infarcted bone → osteomyelitis (Salmonella, Staph. aureus), can be life threatening
• Diagnosis: sickle-shaped RBCs and Howell-Jolly bodies on smear; Hb electrophoresis
• Treatment: hydroxyurea, folic acid; voxelotor (Hgb S polymerization inhibitor) ↓ hemolysis & ↑ Hgb (NEJM 2019;381:509)
• Vaccines: pneumo, meningo, H flu, HBV
• Pain & vaso-occlusive crises: analgesia (consider PCA; ask Pt what worked prev.), IVF, transfusion only if sx & Hgb below Pt’s baseline (often low) given alloimmunization & Fe accumulation (need chelation); Ppx w/ crizanlizumab (anti-P-selectin; NEJM 2017;376:429)
• Acute chest (fever, ↑ WBC, pulm. infilt., r/o other causes): O2, abx, IVF, exchange txfusion
• TIA/stroke: often exchange transfusion (goal Hgb 10) ± thrombolytics
• Gene therapy in development (NEJM 2021;384:205)
Hereditary spherocytosis (HS) (Lancet 2008;372:1411)
• Defect in cytoskeleton of RBC membrane (ankyrin, α- and β-spectrin, band 3, & pallidin)
• Most common in N. European populations (1/5000 births); ⊕ FHx (75% of Pts)
• Anemia, jaundice (mostly neonates), splenomegaly, pigmented gallstones
• Diagnosis: spherocytes on smear, ⊕ osmotic fragility test (~80% Se), ↓ eosin-5-maleimide (EMA) binding (93% Se; 99% Sp; Haemat 2012;97:516), acidified glycerol lysis test (Se 95%)
• Treatment: folate, transfusions, splenectomy for moderate and severe HS (balance w/ ↑ risk of future thrombosis and infection; J Thromb Haemost 2008;6:1289)
Paroxysmal nocturnal hemoglobinuria (see above)
Autoimmune hemolytic anemia (AIHA)
• Acquired, antibody-mediated RBC destruction
• Warm AIHA: IgG Abs opsonize RBCs at body temp → removal by spleen
Etiologies: idiopathic, lymphoproliferative (CLL, NHL), autoimmune (SLE), drugs, HIV, babesiosis (NEJM 2017;376:939)
• Cold AIHA: IgM Ab binds to RBCs at temp <37°C → complement fixation → intravascular hemolysis and acrocyanosis on exposure to cold
Etiologies: idiopathic, lymphoprolif. disorders (eg, Waldenström’s; monoclonal), Mycoplasma pneumoniae infxn and infectious mononucleosis (polyclonal)
• Diagnosis: spherocytes on smear, ⊕ Coombs’; ✓ cold agglutinin titer, splenomegaly
• Treatment (Blood 2017;129:2971): treat underlying disease
Warm AIHA: corticosteroids ± splenectomy, IVIg, cytotoxic agents, rituximab
Cold AIHA: avoid cold; steroids ineffective; rituximab (Blood 2004;103:2925); complement inhibitors (sutimlimab) approved for cold agglutinin disease (NEJM 2021;384:1323)
Drug-induced hemolytic anemia
• Acquired, Ab-drug mediated destruction vs. direct drug effect. Abx: ceph., sulfa drugs, rifampin, ribavirin. CV: methyldopa, procainamide, quinidine, thiazides. TCAs, phenothiazines, NSAIDs, sulfonylureas, MTX, 5-FU, rasburicase (G6PD defic.)
• Diagnosis: Coombs’ usually negative, ↑ LDH; Treatment: discontinue offending agent
Microangiopathic hemolytic anemia (MAHA; NEJM 2014;371:654)
• Intra-arteriolar fibrin damages RBCs → acquired intravascular hemolysis
• Etiologies: hemolytic-uremic syndrome (HUS), thrombotic thrombocytopenic purpura (TTP), disseminated intravascular coagulation (DIC), malignancy, malignant HTN, eclampsia/HELLP, mech. cardiac valves, infected vascular prostheses
• Diagnosis: schistocytes ± ↓ plts ± ↑ PT in DIC, ↑ Cr in HUS, ↑ LFTs in HELLP
• Rx: see “DIC” section in “Coagulopathies” and “TTP/HUS” sections in “Platelet Disorders”
Hypersplenism
• Stasis/trapping in spleen → Mϕ attack/remodel RBCs → spherocytosis → hemolysis
Causes of Splenomegaly |
|
Etiology |
Comments* |
RES hyperplasia |
Hemolytic anemia, sickle cell disease, thalassemia major |
Immune hyperplasia |
Infxn [HIV, EBV, CMV, TB, malaria, kala azar (“black water fever” from visceral leishmaniasis), Mycobacterium avium complex], autoimmune disorders (SLE, RA w/ Felty’s syndrome), sarcoidosis, serum sickness |
Congestion |
Cirrhosis, CHF, portal/splenic vein thrombosis, schistosomiasis |
Infiltration (nonmalignant) |
Lysosomal storage disorders (Gaucher’s, Niemann-Pick), glycogen storage diseases, histiocytosis X, splenic cysts |
Neoplasm |
MPN (CML, PMF, PV, ET), CMML, leukemia, lymphoma (NHL, HL, hairy cell leukemia, CLL, PLL, WM), T-LGL, myeloma, amyloid |
RES = reticuloendothelial system; *boldface = causes of massive splenomegaly.
DISORDERS OF HEMOSTASIS
Clinical Characteristics of Bleeding Disorders |
||
Feature |
Platelet/Vascular Defect |
Coagulation Defect |
Site |
Skin, mucous membranes |
Deep in soft tissues (muscles, joints) |
Lesions |
Petechiae, ecchymoses |
Hemarthroses, hematomas |
Bleeding |
After minor cuts: yes After surgery: immediate, mild |
After minor cuts: unusual After surgery: delayed, severe |
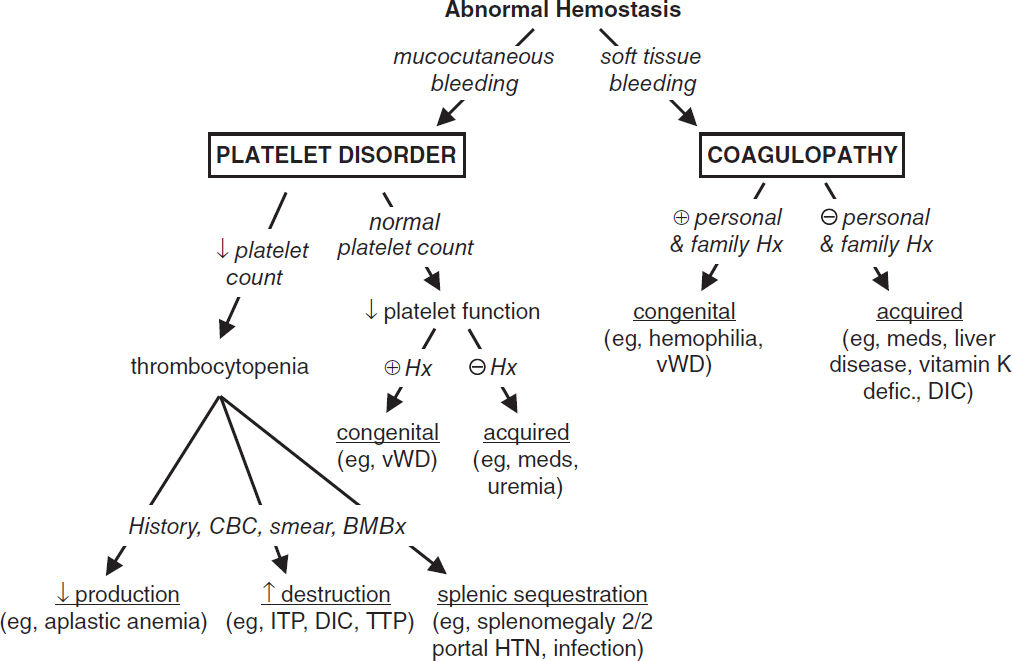
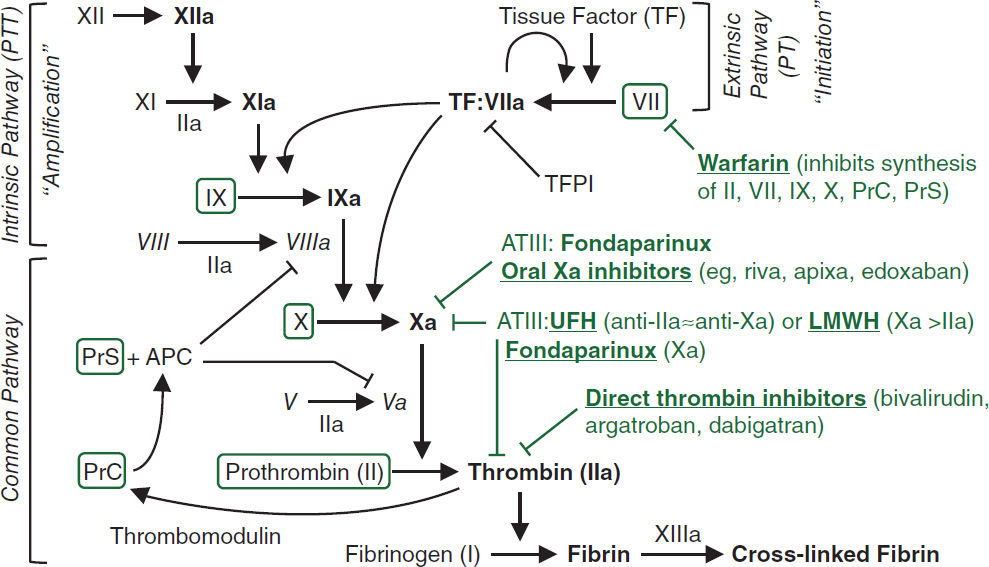
See “Coagulopathies” for reversal agents for anticoagulants. APC, activated protein C; AT, antithrombin; PrC, protein C; PrS, protein S; TF, tissue factor; TFPI, tissue factor pathway inhib.
Purpura (nonblanching purple/red lesions due to extravasation of RBCs into dermis)
• Nonpalpable (macular; ≤3 mm in diameter = petechiae; >3 mm = ecchymoses)
platelet disorder: thrombocytopenia, defect in platelet fxn
thromboemboli: DIC, TTP, cholesterol or fat emboli, other thrombotic microangiopathies
trauma or vascular fragility: amyloidosis, Ehlers-Danlos, scurvy
• Palpable (papular); vasculitis: leukocytoclastic, HSP, PAN, RMSF; infectious emboli: meningococcemia, bacterial endocarditis
• Purpura fulminans (aka retiform purpura): purpura + hypotension + DIC; typically due to infxn/sepsis, protein C or S deficiency or APS (see section on DIC)
PLATELET DISORDERS
THROMBOCYTOPENIA (Plt count <150,000/μL)
Thrombocytopenia and Risk of Bleeding |
|
Platelet Count (cells/µL) |
Risk |
50,000–100,000 |
Risk with major trauma; can proceed with general surgery |
<50,000 |
Risk with minor trauma or surgery |
<20,000 |
Risk of spontaneous bleeding (less so with ITP) |
<10,000 |
Risk of severe, life-threatening bleeding |
Etiologies
• ↓ production
Hypocellular bone marrow: aplastic anemia (qv), rarely MDS, drugs (eg, thiazides, antibiotics, chemotherapy), alcohol, cirrhosis, viral infection
Hypercellular bone marrow: MDS, leukemia, severe megaloblastic anemia
Marrow replacement: myelofibrosis, hematologic and solid malignancies, granulomas
• ↑ destruction
Immune-mediated (distinguish primary from secondary; Blood 2009;113:2386)
1° (idiopathic): immune thrombocytopenic purpura (ITP, see below)
2°: infxn (HIV, HCV, HSV), collagen vascular dis. (SLE), APS, lymphoprolif. (CLL, lymphoma), drugs (many, including heparin, abciximab, quinidine, sulfonamides, vanco), alloimmune (posttransfusion), vaccine-induced
Non–immune-mediated: MAHA (DIC, HUS, TTP), ticlopidine/clopidogrel, vasculitis, preeclampsia/HELLP, cardiopulm bypass, CVVH, IABP, cavernous hemangioma, viral
• Abnormal distribution or pooling: splenic sequestration, dilutional, hypothermia
• Critical illness: multifactorial (Hematology Am Soc Hematol Educ Program 2017;1:660)
• Unknown: ehrlichiosis/anaplasmosis, babesiosis, RMSF
Diagnostic evaluation
• H&P: meds, infxns, underlying conditions, splenomegaly, lymph nodes, bleeding hx
• CBC with differential: isolated thrombocytopenia vs. multilineage involvement
• Peripheral smear (r/o pseudothrombocytopenia due to platelet clumping)
↑ destruction → look for large plts, ↑ MPV, schistocytes (see “Peripheral Smear” inserts)
↓ production → rarely limited to platelets → look for blasts, hypersegmented PMNs, leukoerythroblastic Δs; can see inclusion bodies (anaplasma), parasites (Babesia)
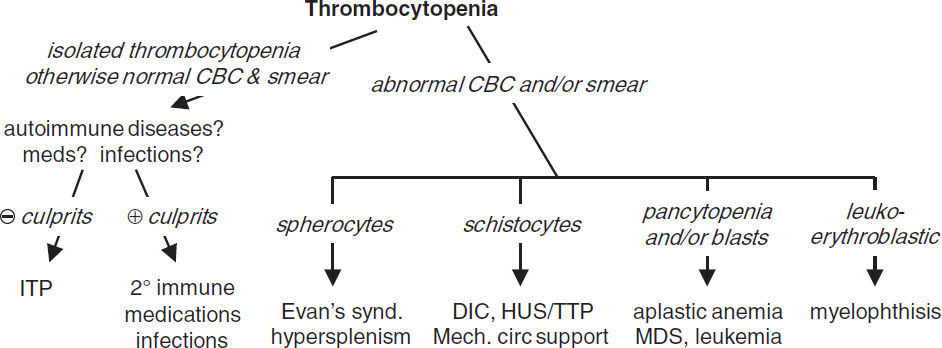
• Additional laboratory evaluations as indicated (eg, viral titers, flow cytometry, ANA, APLA)
if anemia: ✓ reticulocyte count, LDH, haptoglobin, bilirubin to detect hemolysis
if hemolytic anemia: ✓ PT, PTT, fibrinogen, D-dimer, Coombs, ANA
BM bx for unexplained thrombocytopenia, esp. if associated with splenomegaly
Primary immune thrombocytopenic purpura (ITP) (Blood 2010;115:168)
• Isolated thrombocytopenia due to immune plt destruction (auto-Ab to plts) & ↓ production (auto-Ab to megakaryocytes) without precipitant
• Clinical manifestations: insidious onset of mucocutaneous bleeding; ♀:♂ = 3:1
• Diagnosis of exclusion (r/o 2° ITP); no robust clinical or lab parameters, but typically:
CBC: isolated ↓ plt (<100,000/µL); 10% have ITP + AIHA = Evans syndrome
Peripheral smear: large platelets (not specific), r/o pseudothrombocytopenia
BM bx: ↑ megakaryocytes, nl cellularity. Consider if other CBC or smear abnl or diagnostic uncertainty (Blood 2011;117:4910).
✓ HBSAg & anti-HBc prior to rituximab (and before IVIg, which could alter results)
• Treatment: rarely indicated if plt >50,000/µL unless bleeding, trauma/surgery, anticoag.
Treatment of Primary ITP in Adults |
||
Approach |
Treatment |
Notes |
First-line or upfront therapy |
Steroids: prednisone 0.5–2 mg/kg/d PO tapered ~4 wk, or dexamethasone 40 mg PO × 4 d |
↓ Mϕ FcR & ↓ anti-plt Ab 70–90% have initial response ~20% sustained remission |
IVIg (1 g/kg/d IV × 2–3 d) Consider if need rapid ↑ in plt in 24–48 hrs; lasts 2–6 wks |
Blocks Mϕ FcR, ↓ anti-plt Ab Interferes w/ Mϕ uptake Ab-coated plts; 80% have initial response |
|
Anti-Rh(D) Ig: alternative to IVIg if RBC Rh(D) ⊕; 50–75 mcg/kg/d |
Ab-coated RBCs overwhelm Mϕ FcR Avoid if h/o hemolysis; not often used |
|
Second-line or maint. therapy |
Romiplostim, el/avatrombopag |
TPO-R agonists → ↑ plt prod |
Rituximab (anti-CD20) ± dex |
anti–B-cell Ab |
|
Splenectomy* (less common) |
~65% long-term remission |
|
AZA, CYC, MMF |
Immunosuppressants |
|
Danazol: 600 mg/d |
Androgen (hirsutism) ↓ plt clearance |
|
Chronic/ refractory |
Romiplostim or eltrombopag |
Allows splenectomy to be deferred |
Fostamatinib: 75–150 mg BID |
Spleen tyrosine kinase (SYK) inhibitor |
|
Vinca alkaloids |
Good initial response, less durable |
|
Bleeding |
Aminocaproic acid |
Inhibits plasmin activation |
Methylprednisolone 1 g/d IV × 3 d |
See above |
|
IVIg |
See above |
|
Platelet transfusion |
Given w/ IVIg or anti-Rh(D) |
|
*Post-splenectomy vaccinations needed. (Blood Adv 2019;3:3829; Eur J Haem 2018;100:304)
Secondary immune thrombocytopenic purpura (2° ITP)
• Diagnosis: viral serologies (HIV, HCV, HBV, EBV), H. pylori Ab, ANA, pregnancy test, APLA, TSH, parvovirus, & CMV PCR. Anti-plt Ab tests often sent but less useful.
Heparin-Induced Thrombocytopenias (Chest 2012;141:e495S; NEJM 2015;373:252) |
||
Feature |
Type I (not clin. signif) |
Type II (clinically significant HIT) |
Mechanism |
Direct effect of heparin (non-immune) |
Immune (Ab)-mediated IgG against plt factor 4—heparin complex |
Incidence |
10–20% |
1–3% with UFH, 0–0.8% LMWH |
Onset |
After 1–4 d of heparin therapy |
After 4–10 d, but can occur in <24 h if prior exposure w/in 100 d (persistent Ab). Postop highest risk. Can occur after heparin d/c. |
Platelet nadir |
>100,000/µL |
~60,000/µL, ↓ >50% |
Sequelae |
None |
Thrombotic events (HITT) in 30–50% |
Management |
Can continue heparin and observe |
Discontinue heparin Alternative anticoagulation |
• Treat underlying etiology
• Pathophysiology (type II): Ab binds heparin-PF4 → immune complex binds to plt → plt activation, further PF4 release → plt aggregates removed from circulation → thrombocytopenia; procoagulants released by plts and tissue factor released by endothelial cells damaged by HIT Abs → prothrombotic state
• Diagnosis (need clinical + pathologic)
Clinical: plt <100k or ↓ 50% from baseline; or venous (DVT/PE) or arterial (limb ischemia, CVA, MI) thrombosis (4:1 ratio); skin necrosis; ? ↑ heparin resistance
Pathologic: ⊕ HIT Ab using PF4-heparin ELISA (≥90% Se, IgG-specific ELISA Sp 94%), may confirm w/ functional plt assay (serotonin-release) (>95% Se/Sp)
Clinical context important: HIT Ab (esp. IgM ELISA) may be ⊕ in 10–20% of Pts on UFH/LMWH (Am J Hem 1996;52:90), up to 50% of cardiac bypass Pts (Circ 1997;95:1242)
Pretest prob w/ “4 T’s” criteria (Blood 2012;120:4160): ≤3 points → 99% NPV, investigate other causes; 4–5 points 22% PPV & 6–8 points 64% PPV, ✓ lab test & replace UFH
Evaluation of Suspected HIT (“4T’s”) |
|||
Factor |
2 Points |
1 Point |
0 Points |
Thrombo-cytopenia |
↓ >50% and nadir ≥20k |
↓ 30–50% or nadir 10–19k |
↓ <30% or nadir <10k |
Timing |
5–10 d or ≤1 d if heparin w/in 30 d |
? 5–10 d (but not clear), >10 d or ≤1 d if hep w/in 30–100 d |
≤4 d w/o recent hep |
Thrombosis |
New thromb, skin necrosis, acute rxn after IV UFH |
Prog/recurrent thromb, suspect thromb or non-nec skin lesion |
None |
Other cause |
None apparent |
Possible |
Definite |
• Treatment of HIT (type II) (NEJM 2015;373:252; Blood Adv 2018;2:3360)
Discontinue heparin (incl. flushes, LMWH Ppx, heparin lines). Avoid plts (anecdotal link w/ thrombosis); if given warfarin, give vit K to reverse, prevent warfarin skin necrosis.
Nonheparin anticoag (argatroban, bivalirudin; NEJM 2013;368:737) regardless of thrombosis; start warfarin when plt >150k, overlap ≥5 d or DOAC (Blood 2017;130:1104)
⊕ thrombosis (HITT): anticoagulate for ≥3–6 mo
⊖ thrombosis (HIT): screen for DVT; unclear duration of subsequent anticoag (until plt count recovers, often ~2–3 mo if no clot); 25–50% thrombosis rate w/in 30 d
• H/o HIT: if PF4 Ab ⊖ or SRA ⊖ (typically >100 d after dx) → may consider re-exposure to UFH (eg, for surgery); HIT recurrence low but can be seen (Blood 2014;123:2485)
Thrombotic microangiopathies (TMA; NEJM 2014;371:654; Lancet 2017;390:681)
• Endothelial injury → plt aggreg. & microvasc. thrombosis → ↓ plt & RBC hemolysis (MAHA)
• Dx: unexplained thrombocytopenia (typically <20k) + MAHA → sufficient for dx ⊕ schistocytes (>2–3/hpf), ⊖ Coombs, normal PT/PTT & fibrinogen ↑↑ LDH (tissue ischemia + hemolysis), ↑ indirect bili., ↓↓ haptoglobin, ↑ Cr (esp. in HUS)
Biopsy: arterioles filled with platelet hyaline thrombi
Ddx: DIC, vasculitis, malignant hypertension, preeclampsia/HELLP syndrome
• Thrombotic thrombocytopenic purpura (TTP)
Pathophys: ↓↓ ADAMTS13 protease activity (hereditary [Upshaw Schulman Syn.] or autoAb) → vWF multimers persist on endothelial surface → plt adhesion/aggregation → thrombosis
Clinical: pentad (all 5 in only ~5%) = ↓ plts + MAHA (100%) ± Δ MS (65%) ± renal failure (50%, late feature) ± fever (25%)
PLASMIC score to discriminate TTP from other TMAs (Lancet Haematol. 2017;4:157)
Rx: urgent plasma exchange ± glucocorticoids; FFP if delay to plasma exchange, caplacizumab (NEJM 2019;380:335), rituximab for 2° prevention (Blood Adv. 2017;1:1159),
plt transfusions contraindic. → ↑ microvascular thromb (J Thromb Haemost. 2020;18:2496)
• Hemolytic-uremic syndrome (HUS)
Pathophys: (1) Shiga toxin damages renal endothelial cells → intrarenal thrombi; or (2) complement dysregulation (hereditary or acquired), so-called “atypical HUS”
Clinical: triad = thrombocytopenia + MAHA + renal failure (bloody diarrhea if Shiga)
Rx: supportive care; eculizumab (J Nephrol 2017;30:127); plasma exchange if CNS sx
• Drug-induced TMA (clinically similar to TTP; Blood 2017;129:2857)
Immune-mediated (Ab reacts w/ plts & endothelial cells): eg, quinine, gemcitabine?
Direct toxicity mediated: eg, gemcitabine, mitomycin, tacrolimus, CsA, bevacizumab
Disseminated intravascular coagulation (DIC): see “Coagulopathies”
DISORDERS OF PLATELET FUNCTION
Mechanisms and Etiologies of Platelet Function Abnormalities |
||
Function |
Inherited |
Acquired |
Adhesion |
Bernard-Soulier; vWD |
Uremia; acquired vWD |
Aggregation |
Afibrinogenemia Glanzmann’s thrombasthenia |
P2Y12 inhibitors, GP IIb/IIIa inhibitors Dysproteinemias (myeloma) |
Granule release |
Chediak-Higashi syndrome Hermansky-Pudlak syndrome |
Drugs (ASA, NSAIDs); liver disease; MPN; cardiopulmonary bypass |
Tests of platelet function
• Platelet aggregation tests: measure aggregation in response to agonists (eg, ADP)
von Willebrand’s disease (vWD) (NEJM 2016;375:2067)
• von Willebrand’s factor (vWF) function = platelet glue & plasma carrier of factor VIII
• vWD most common inherited bleeding disorder; ~85% (type 1) have partial quantitative vWF defic., ~15% (type 2) qualitative defic., <1% (type 3) total/near-total absence of vWF
• Acquired vWD: a/w many disorders (malig, MPN w/ ↑ plt count; autoimmune; hypo-thyroidism; drugs) and caused by different mechanisms (anti-vWF Abs, ↑ clearance, ↓ synthesis); Heyde’s syndrome = vWF destruction by severe AS, a/w GI AVMs/bleed
• Diagnosis: ↓ vWF:Ag, ↓ vWF activity (measured by ristocetin cofactor assay), ↓ factor VIII, ± ↑ PTT, ± ↓ platelets; confirm with vWF multimer analysis
• Clinical condition, factor VIII levels and ristocetin cofactor assay useful to guide Rx decision
• Rx: desmopressin (dDAVP, IV/IN; tachyphylaxis) → ↑ endothelial cell release of vWF; efficacy depends on type (avoid in type 2), ∴ ✓ response before use w/ bleeding or procedures; vWF replacement: cryo, vWF-rich factor VIII concentrates, recomb. vWF
Uremic bleeding
• Uremia → platelet dysfunction including ↓ aggregation, impaired adhesiveness
• Treatment: dDAVP, cryoprecipitate, correct anemia (improves plt aggregation and adhesion by increasing plt interactions with endothelium), consider holding anti-plt agents
COAGULOPATHIES
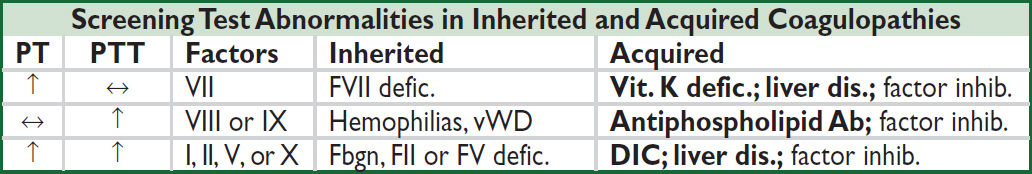
Further coagulation tests (JAMA 2016;316:2146)
• Mixing study: useful if ↑ PT or PTT; mix Pt’s plasma 1:1 w/ normal plasma and retest
PT/PTT normalizes → factor deficiency; PT/PTT remains elevated → factor inhibitor
• Coagulation factor levels: useful if mixing study suggests factor deficiency
DIC → all factors consumed; ∴ ↓ factors V and VIII
Liver disease → ↓ all factors except VIII; ∴↓ factor V, normal factor VIII
Vitamin K deficiency → ↓ factors II, VII, IX, X (and protein C, S); ∴ normal V and VIII
• DIC screen: ↓ fibrinogen (consumed), fibrin degradation products (FDPs, ⊕ from intense fibrinolysis), ↑ D-dimer (more specific FDP test that detects degradation of X-linked fibrin)
Hemophilias (Lancet 2016;388:187)
• X-linked recessive factor VIII (hemophilia A) or factor IX (hemophilia B) deficiency
• Classification: mild (5–25% normal factor activity), moderate (1–5%), or severe (<1%)
• Clinical manifestations: hematomas, hemarthroses, bruising, bleeding (mucosal, GI, GU)
• Diagnosis: ↑ PTT (normalizes w/mixing study), normal PT & vWF, ↓ factor VIII or IX
• Prophylaxis indicated if <1% activity of factor VIII or IX
• Rx: purified/recomb. factor VIII or IX; desmopressin (mild disease); anti-fibrinolytics (aminocaproic acid; tranexamic acid); cryo (FVIII); emicizumab (bridges factor IX and X), effective for hemophilia A w/ and w/o inhibitors (NEJM 2017;377:809 & 2018;379:811)
Coagulation factor inhibitors (anti-factor antibodies; anti-factor VIII most common)
• Etiologies: hemophilia; postpartum; lymphoproliferative & autoimmune disorders; cancers
• Diagnosis: ↑ PTT (does not normalize w/ mixing study); Bethesda assay quantitates titer
• Rx: if high titer → recomb. factor VIIa, porcine factor concentrates, activated prothrombin complex; for others → high-purity human factor, plasmapheresis, immunosuppression
Disseminated intravascular coagulation (DIC) (NEJM 2014;370:847)
• Etiologies: trauma, shock, infection, malignancy (esp. APL), obstetric complications
• Pathogenesis: massive activation of coagulation that overwhelms control mechanisms
Thrombosis in microvasculature → ischemia + microangiopathic hemolytic anemia
Acute consumption of coagulation factors and platelets → bleeding
Chronic DIC → able to compensate by ↑ factors and platelets → thrombosis
• Diagnosis: ↑ PT, ↑ PTT, ↓ fibrinogen (may be nl b/c acute phase), ⊕ FDP/D-dimer, ↓ plts, ⊕ schistos, ↑ LDH, ↓ hapto; chronic DIC: ⊕ FDP/D-dimer, variable plts, other labs nl
• Treatment: Rx underlying process; support w/ FFP, cryo (goal fbgn >100 mg/dL) & plts
Vitamin K deficiency
• Etiologies: malnutrition, ↓ absorption (antibiotic suppression of vitamin K-producing intestinal flora or malabsorption), liver disease (↓ stores), warfarin
Properties and Antidotes for Anticoagulants & Fibrinolytics (Circ 2016;134:248) |
|||
Anticoag. |
t1/2 |
Labs |
Rx for O/D w/ Serious Bleeding (+ d/c anticoag) |
UFH |
60–90′, RES |
↑ PTT |
Protamine IV 1 mg/100 U UFH (max 50 mg). For infusions, dose to reverse 2× UFH given per h. |
LMWH |
2–7°, K |
anti-Xa* |
Protamine reverses ~60%. 1 mg per 1 mg enox. |
Bivalirudin |
25′, K |
↑ PTT |
Dialysis |
Argatroban |
45′, L |
↑ PTT |
? Dialysis |
Warfarin |
36°, L |
↑ PT |
No bleeding: vit K 2.5 mg PO if INR >9, o/w no e/o clinical benefit (Blood Adv 2019;3:789) Bleeding: vit. K 10 mg IV + either 4F-PCC (KCentra, 25, 35, or 50 U/kg for INR 2–4, 4–6, or >6) or FFP 2–4 U IV q6–8h (slower, more volume) |
Fibrinolytic |
20′, LK |
↓ fbgn |
Cryo, FFP, ± tranexamic or aminocaproic acid |
Rivaroxaban Apixaban Edoxaban |
8–12°, K >L |
↑ PT* anti-Xa* |
Andexanet alfa (factor Xa decoy receptor): 800 mg bolus (30 mg/min) → 8 mg/min infusion (½ of above if taking ½ dose DOAC or ≥8 hrs since last dose; NEJM 2019;380:1326); 4F-PCC if andexanet not available |
Dabigatran |
~12°, K |
↑ PTT* |
Idarucizumab: mAb binds drug (NEJM 2017;377:431) |
*Routine monitoring not performed. Mode of excretion: K, kidney; L, liver; RES, reticuloendothelial system. 4F-PCC: prothrombin complex concentrate (FII, VII, IX, X; Protein C & S). Anti-fibrinolytics: tranexamic, aminocaproic acid.
HYPERCOAGULABLE STATES
Suspect in Pts with venous or arterial thrombosis at young age or unusual locations, recurrent thromboses or pregnancy loss, or ⊕ FHx
Inherited Hypercoagulable States |
|||
Risk Factor |
Prevalence |
VTE* |
Comments |
Factor V Leiden |
3–7% |
2.65 |
Activated protein C (APC) resistance |
Prothrombin mutation |
2% |
1.45 |
G20210A → ↑ prothrombin level |
Hyperhomocysteinemia |
5–10% |
_ |
Inherited or acquired (vitamin defic., hypothyroid, renal insufficiency) |
Protein C deficiency |
0.02–0.05% |
2.8 |
Warfarin-induced skin necrosis risk |
Protein S deficiency |
0.01–1% |
2.8 |
|
Antithrombin III def. |
0.04% |
2.8 |
May be heparin resistant |
*Relative risk of recurrent VTE compared to patient w/o respective thrombophilia (JAMA 2009;301:2472)
Vascular Beds Affected by Inherited and Acquired Hypercoagulable States |
||
|
Venous |
Venous and Arterial |
Inher. |
Factor V Leiden Prothrombin mutation ↓ protein C, S or AT III |
Hyperhomocysteinemia (inherited or acquired) Dysfibrinogenemia |
Acquired |
Stasis: immobilization, surgery, CHF Malignancy Hormonal: OCPs, HRT, tamoxifen, pregnancy Nephrotic syndrome |
Platelet defects: myeloproliferative disorders, HIT, PNH (although venous >arterial) Hyperviscosity: polycythemia vera, Waldenström’s -macroglobulinemia, sickle cell, acute leukemia Vessel wall defects: vasculitis, trauma, foreign bodies Others: antiphospholipid syndrome, IBD |
Diagnostic evaluation (not routinely required for initial VTE; NEJM 2017;377:1177)
• APC resistance screen; prothrombin PCR test; functional assays for proteins C and S, ATIII; homocysteine level; factor VIII levels; anticardiolipin and lupus anticoagulant Ab. Also consider nephrotic syndrome, PNH (esp. if mesenteric thrombus).
• Consider JAK2 mutation testing if suspect MPN or splanchnic thrombosis
• Proteins C & S and ATIII levels unreliable during acute thrombosis and anticoagulation ∴ levels best assessed ≥2 wk after completing anticoagulation course
• Age-appropriate malignancy screening (occult cancer in ~4% of initial unprovoked VTE; no benefit of routine abd/pelvis CT; NEJM 2015;373:697)
Treatment
• Asx w/ inherited risk factor: consider prophylactic anticoag. if develops acquired risk factor
• Thrombosis w/ inherited risk factor: see “Venous Thromboembolism”
Antiphospholipid syndrome (APS) (NEJM 2018;398:2010)
• Definition: dx requires ≥1 clinical & ≥1 laboratory criteria
Clinical: thrombosis (any) or complication of pregnancy (≥3 spont. abortions before 10 wk or ≥1 fetal loss after 10 wk or premature birth before 34 wk)
Laboratory: ⊕ lupus anticoagulant (LA), or ⊕ moderate–high titer anticardiolipin (ACL), or ⊕ β2-glycoprotein-I (β2-GP-I) Ab, on ≥2 occasions, at least 12 wk apart
• Features: DVT/PE/CVA, recurrent fetal loss, ↓ plts, hemolytic anemia, livedo reticularis
• “Catastrophic APS”: ≥3 organ systems in <1 wk w/ ⊕ APLA & tissue microthrombi; 44% mortality (Arth Rheum 2006;54:2568); Rx w/ plasmapheresis, rituximab
• Antiphospholipid antibodies (APLA)
✓ if: SLE, age <40 y & arterial thromb, recurrent venous thromb, spontaneous abortion
ACL: Ab against cardiolipin, a mitochondrial phospholipid; IgG more specific than IgM
LA: Ab that prolongs phospholipid-dependent coagulation reactions; ∴ ↑ PTT that does not correct with mixing study but does correct with excess phospholipids or platelets; PT not affected b/c the reaction contains much more phospholipid
β2-GP-I: Ab against β2-glycoprotein-I, IgG or IgM (uncertain role of Abs in pathogenesis)
False ⊕ VDRL nontreponemal test for syphilis (cardiolipin is part of Ag complex)
Risk of thromboembolic phenomena may increase with titer of APLs
• Etiologies: primary (idiopathic) or secondary due to autoimmune syndromes (eg, SLE), malignancy, infections, drug reactions
• Treatment: UFH/LMWH → warfarin (lifelong for most Pts)
Rivaroxaban inferior to warfarin in triple positive (⊕ ACL, LA, & β2-GP) (Blood 2018;132:1365)
Initial venous thrombosis: INR 2–3 (NEJM 2003;349:1133; J Thromb Haemost 2005;3:848)
Initial arterial thrombosis: typically INR 2–3 + ASA 81 mg/d
Recurrent thrombosis on warfarin: consider INR 3–4 vs. LMWH or fondaparinux
DISORDERS OF LEUKOCYTES
Neutrophilia (>7500–10,000/µL) |
|
Infection |
Usually bacterial; ± toxic granulations, Döhle bodies |
Inflammation |
Burn, tissue necrosis, MI, PE, collagen vascular disease |
Drugs and toxins |
Corticosteroids, β-agonists, lithium, G-CSF; cigarette smoking |
Stress |
Release of endogenous glucocorticoids and catecholamines |
Marrow stimulation |
Hemolytic anemia, immune thrombocytopenia |
Asplenia |
Surgical, acquired (sickle cell), congenital (dextrocardia) |
Neoplasm |
Can be 1° (MPN) or paraneoplastic (eg, carcinomas of lung, GI) |
Leukemoid reaction |
>50,000/µL + left shift, not due to leukemia; unlike CML, ↑ LAP |
Neutropenia (<1000/µL) |
|
Congenital |
Myelokathexis, Shwachman-Diamond-Oski, Chédiak-Higashi, retic dysgen., WHIM syndrome, cyclic neutropenia, monoMAC syndrome (↓ monos, NKs) |
Infection |
Viral (CMV, EBV, HIV); bacterial (Brucella, Rickettsia, TB); malaria |
Nutritional |
Vitamin B12 or copper deficiency |
Drugs and toxins |
Chemotherapeutics, clozapine, methimazole, TMP-SMX, NSAIDs, sulfasalazine, phenytoin (Am J Hem 2009;84:428), alcohol |
Neoplasm |
MDS, leukemia (AML, ALL, hairy cell, LGL, others) |
Monocytosis (>500/µL) |
|
Infection |
Usually TB, SBE, Listeria, Brucella, Rickettsia, fungi, parasites, syphilis |
Inflammation |
IBD, sarcoidosis, collagen vascular diseases |
Stress |
MI, splenectomy, exercise (Cytokine 2013;61:364) |
Neoplasm |
Hodgkin lymphoma, leukemias, MPNs, carcinomas |
Eosinophilia (>500/µL) |
|
Infection |
Usually parasitic (helminths) |
Allergic |
Drugs; asthma, hay fever, eczema; ABPA |
Collagen vasc dis. |
RA, EGPA (Churg-Strauss), eosinophilic fasciitis, PAN |
Endocrine |
Adrenal insufficiency |
Neoplasm |
HL, CML, mycosis fungoides, carcinomas, systemic mastocytosis |
Atheroembolic dis. |
Cholesterol emboli syndrome |
Hypereosinophilic syndrome |
Multiorgan involvement incl. heart & CNS, a/w FIP1L1-PDGFRA fusion (NEJM 2003;348:1201); often steroid resistant |
Basophilia (>200/µL) |
|
Neoplasm |
MPN, CML, AML, Hodgkin lymph. |
Infection |
TB, smallpox, parasites |
Alteration in BM or reticuloendothelial compartment |
Hemolytic anemia, splenectomy |
Inflammation or allergy |
IBD, chronic airway inflammation |
Lymphocytosis (>4000–5000/µL) |
|
Infection |
Usually viral; “atypical lymphocytes” with mononucleosis syndromes Other: pertussis, toxoplasmosis |
Hypersensitivity |
Drug-induced, serum sickness |
Autoimmune |
Rheumatoid arthritis (large granular lymphocytes), malignant thymoma |
Neoplasm |
Leukemia (eg, CLL, hairy cell, LGL), lymphoma (eg, mantle cell, folic.) |
Lymphadenopathy |
|
Viral |
HIV, EBV, CMV, HSV, VZV, hepatitis, measles, rubella |
Bacterial |
Generalized (brucellosis, leptospirosis, TB, atypical mycobacteria, syphilis) Localized (streptococci, staphylococci, cat-scratch disease, tularemia) |
Fungal/parasitic |
Histo, coccidio, paracoccidioidomycosis, toxoplasmosis |
Immunologic |
Collagen vascular disease, drugs (eg, phenytoin), serum sickness, histiocytosis X, Castleman’s and Kawasaki disease |
Neoplasm |
Lymphoma, leukemia, amyloidosis, metastatic carcinoma |
Other |
Sarcoidosis; lipid storage diseases |
Factors that favor biopsy |
Pt >40 y, >2 cm, location (supraclavicular always abnl), duration >1 mo Consistency (hard vs. rubbery vs. soft) & tenderness are not reliable Exicisional biopsy preferred over fine needle aspiration (FNA) |
TRANSFUSION THERAPY
Blood Products and Indications (Lancet 2013;381:1845) |
|||
Packed red blood cells (PRBCs) |
For acute blood loss or to ↑ O2-carrying capacity if end organ ischemia. 1 U PRBC → ↑ Hb by ~1 g/dL. Hb goal >7 g/dL adequate for UGIB & critically ill (NEJM 2013;368:11 & 2014;371:1381), ≥8 in acute MI and peri-cardiac surgery (NEJM 2018;379;1224; JAMA 2021;325:552). |
||
Platelets (plts) (Annals Int Med 2015;162:205) |
For plts <10k (NEJM 2010;362:600) due to chemo/abx. If <20k or if <50k w/ active bleeding. Variable pre-procedure.100k for neurosurgery. 1 U → ↑ plt ~30–60k. Single donor plt apheresis ↓ alloimmunization. Contraindic: TTP/HUS, HELLP, HIT. Refractory if ↑ <5k 30–60′ post-plts. Suggests consumption such as ITP, DIC, or alloimmunization → trial ABO-matched plts & give more. If still refractory ✓ panel reactive Abs to assess utility of HLA-matched plts. |
||
Fresh frozen plasma (FFP) |
Contains all coagulation factors. For bleeding due to defic. of multiple coag factors (eg, DIC, TTP/HUS, liver disease, dilution). Nb, reverse warfarin w/ Kcentra = 4 factor PCC (JACC 2020;76:594). |
||
Cryoprecipitate |
Enriched for fibrinogen, vWF, VIII, and XIII. 1st line for fibrinogen <100 mg/dL. For bleeding in vWD factor XIII deficiency, use if other products not available. |
||
Irradiated |
Prevents donor T-cell engraftment and risk of transfusion-assoc. GVHD (HSCT, heme malignancy, congenital immunodeficiency). |
||
CMV-negative |
From CMV-negative donors. For CMV-seronegative pregnant women, transplant candidates/recipients, SCID, AIDS Pts. |
||
Leuko- reduced |
WBCs cause HLA alloimmunization & fever (cytokines) and carry CMV. For chronically transfused Pts, potential Tx recip., h/o febrile nonhemolytic transfusion rxn, cases in which CMV-neg products desired but unavailable. |
||
IV immune globulin (IVIg) |
Polyvalent IgG from >1000 donors. For postexposure prophylaxis (eg, HAV), certain autoimmune disorders (eg, ITP, Guillain-Barré, MG, CIDP), congenital or acquired hypogammaglobulinemia (CVID, CLL). |
||
Therapeutic apheresis |
Removes plasma large molec wt subst. (eg, cryoglobulinemia, Goodpasture’s, Guillain-Barré, hyperviscosity syndrome), or cells (eg, leukemia w/ hyperleukocytosis, sx thrombocytosis) from plasma. TTP: replace ADAMTS13. RBC exchange for SCD acute chest or stroke. |
||
Massive transfusion |
Large-vol. PRBC → ↓ Ca, ↑ K, ↓ plt, ↑ coags; initial ratio of 1 PRBC: 1 plt:1 FFP accepted but controversial, follow labs (JAMA Surg 2017;152:574). |
||
Transfusion Complications (NEJM 2017;377:1261) |
|||
Noninfectious |
Risk (per unit) |
Infectious |
Risk (per unit) |
Febrile |
1:100 |
CMV |
Common |
Allergic |
1:100 |
Hepatitis B |
1:220,000 |
Delayed hemolytic |
1:50–75,000 |
Hepatitis C |
1:1,600,000 |
Acute hemolytic |
1:200,000 |
HIV |
1:1,800,000 |
Febr. non-hemolytic |
1:200 |
Bacteria (PRBCs) |
1:500,000 |
TRALI |
1:5000 |
Bacteria (platelets) |
1:12,000 |
Transfusion reactions
• Reason why blood products (unless massive txfusion) run 1 at a time. For all rxns (except minor allergic): stop txfusion; send remaining blood product + fresh blood draw to blood bank.
• Acute hemolytic: fever, HoTN, flank pain, AKI w/in 24 h. Due to ABO incompatibility → preformed Abs vs. donor RBCs. Rx: IVF, ↑ UOP w/ diuretics, mannitol, or dopamine.
• Delayed hemolytic: generally less severe than acute hemolytic; 5–7 d after transfusion, but can be severe → hyperhemolysis. Due to undetected allo-Abs vs. minor antigens → anamnestic response. Rx usually not required; dx important for future transfusion.
• Febrile nonhemolytic: fever, rigors 0–6 h post transfusion. Due to Abs vs. donor WBCs and cytokines in blood product. Rx: acetaminophen ± meperidine; r/o infection, hemolysis.
• Allergic: urticaria; rarely, anaphylaxis due to rxn to txfused proteins, especially in IgA-deficient Pts w/ anti-IgA Abs (use washed products). Rx: urticaria → diphenhydramine; anaphylaxis → epinephrine ± steroids. Washed products ↓ rxns in chronic txfusions.
• Transfusion-associated circulatory overload (TACO): ↑ volume → pulm. edema, ↑ BP. Rx: slow transfusion rate, diuretics, O2, ± nitrates, ± positive pressure ventilation.
• Transfusion-related acute lung injury (TRALI): non-cardiogenic pulm. edema due to donor allo-Abs (from multiparous ♀ plasma) binding recipient WBCs, which then aggregate in pulmonary vasculature and release mediators causing ↑ capillary permeability. Rx: see “ARDS.” No longer seen in US as plasma only from ♂ donors.
MYELODYSPLASTIC SYNDROMES (MDS)
Myeloid neoplasm overview (Blood 2016;127:2391)
• Categories based on clinical features, morphology, immunophenotyping, and genetics
WHO 2016 Classification of Myeloid Neoplasms & Acute Leukemia |
|
Acute myeloid leukemia |
Clonal myeloid stem cell (SC) disorder w/ ≥20% blasts |
Myelodysplastic syndromes |
Dysplastic clonal myeloid SC disorder → cytopenias; <20% blasts, risk of leukemic transformation |
Myeloproliferative neoplasms |
Nondysplastic multipotent myeloid SC clonal expansion |
MDS/MPN neoplasms |
Features of MDS & MPN (eg, CMML, atypical CML) |
Myeloid/lymphoid malig. w/ eos & rearrangements of PDGFR or FGFR1 or w/ PCM1-JAK2 |
May be responsive to TKI therapy (eg, imatinib) for PDGFR rearrangement |
Mastocytosis |
Clonal mast cell disorder, assoc w/ KIT mutations |
Myeloid neoplasms w/ germ line predisposition |
MDS, MDS/MPN, acute leukemias in background of predisposing germline mutations (eg, DDX41) |
Myelodysplastic syndromes (MDS) overview (Lancet 2014;383:2239)
• Acquired clonal stem cell disorder → ineffective hematopoiesis → cytopenias, dysmorphic blood cells and precursors, variable risk of leukemic transformation
• Epidemiology: 20–30,000 cases/y; median age ~70 y; male predominance (1.8×)
• Idiopathic or 2° to chemo w/ alkylating agents; ↑ risk w/ radiation, benzene
• Clinical manifestations: anemia (85%), neutropenia (50%), thrombocytopenia (40–65%)
• Diagnosis: dysplasia (usually multilineage) in peripheral smear (oval macrocytes, pseudo-Pelger-Huët anomaly) and bone marrow (≥10% dysplasia with blasts ± RS)
• Both cytogenetic [eg, del(5q), mono 7, del(7q), trisomy 8, del(20q)] and molecular abnl (TP53, EZH2, ETV6, RUNX1, ASXL1, SF3B1, DNMT3A) may carry prognostic signif
• Prior to dx MDS: exclude AML (≥20% blasts) and CMML (monos >1 × 109/L); r/o 2° BM Δs (defic. of B12, folate, copper); viral infxn (eg, HIV); EtOH; lead, arsenic exposures
WHO 2016 Classification Systems for MDS (Blood 2016;127:2391) |
||
Classification |
WHO 2008 |
Features |
MDS w/ single lineage dysplasia (MDS-SLD) |
RCUD (RA/RN/RT) |
1 dysplastic lineage, 1–2 cytopenias, <15% RS*, <5% BM/ <1% PB blasts, no Auer rods |
MDS w/ multilineage dysplasia (MDS-MLD) |
RCMD |
2–3 dysplastic lineages, 1–3 cytopenias, <15% RS*, <5% BM/ <1% PB blasts, no Auer rods |
MDS w/ ring sideroblast (MDS-RS) |
RARS |
≥15% RS or ≥5% RS if SF3B1 mut. is present, <5% BM/<1% PB blasts, no Auer rods |
MDS w/ isolated del(5q) |
Del(5q) |
Del(5q) alone or w/ 1 abnl except –7 or del(7q) |
MDS w/ excess blasts (MDS-EB) |
RAEB-1 RAEB-2 |
EB-1: 5–9% BM/2–4% PB blasts, no Auer rods EB-2: 10–19% BM/5–19% PB blasts or Auer rods |
MDS, unclassifiable (MDS-U) |
MDS-U |
w/ 1% PB blasts, single lineage dysplasia & pancytopenia, or defining cytogenetic alteration |
Certain cytogenetics [eg, t(15;17), t(8;21), inv16, t(16;16), or MLL rearrangement] classified as AML, regardless of BM blast count. BM, bone marrow; PB, peripheral blood; RS, ring sideroblast. * <5% RS if SF3B1 mutation.
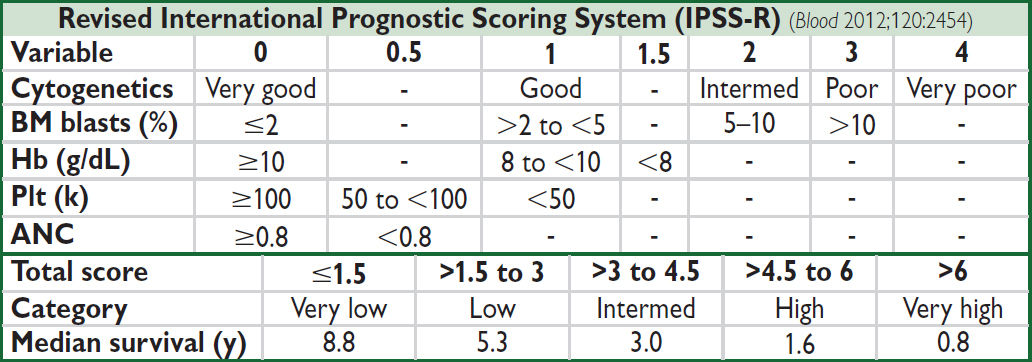
• Rx (Am J Hematol 2012;87:692): intensity based on IPSS-R (qv), age, performance status (PS)
Poor PS, any risk → supportive care (transfusions, G-CSF, Epo, TPO-mimetic, abx prn)
Very low/low risk (IPSS-R) → ESA if Epo <500; lenalidomide (esp. for 5q synd.; Blood 2011;118:3765); luspatercept (MDS-RS). TPO-mimetic agents, transfusions.
Interm/high risk (IPSS-R) → allogeneic HSCT if medically fit, DNA methyltransferase inh azacitadine or decitabine (Lancet Oncol 2009;10:223) or oral decitabine/cedazuridine
Hypoplastic MDS (rare) → consider immunosuppression (CsA, ATG), HSCT
• Prognosis: mutations inTP53, ASXL1, EZH2, RUNX1, ETV6 (NEJM 2011; 364:2496) ↓ survival
MYELOPROLIFERATIVE NEOPLASMS (MPN)
General (NEJM 2017;376:2168)
• Results from clonal expansion of multipotent hematopoietic stem cell
• Categories of MPN: polycythemia vera (PV); essential thrombocythemia (ET); primary myelofibrosis (PMF); chronic myelogenous leukemia (CML; BCR-ABL1 ⊕); atypical CML (aCML); chronic neutrophilic leukemia (CNL); systemic mastocytosis; chronic eosinophilic leukemia; MPN-NOS/unclassifiable
• MDS/MPN neoplasms: proliferative and dysplastic features
• Mutations useful as clonal markers & dx tools:
Gain of fxn mutations in JAK2 V617F (Janus kinase) frequently present (PV ~95%, ET ~50%, PMF ~50%; NEJM 2005;352:1779)
BCR-ABL1 fusion in all cases of CML; SETBP1 in aCML
CALR exon 9 mutation, type I and II (most MPNs w/o JAK2 or MPL mutation, including ~25% of ET, ~35% of myelofibrosis Pts; NEJM 2013;369:2379 & 2391)
Type I has better prognosis.
MPL, TET2, & ASXL1 mutation w/ lower frequency
CSF3R mutation present in ~60% of CNL; KIT D816V in 90% of systemic mastocytosis
POLYCYTHEMIA VERA (PV)
Definition
• ↑ in RBC mass ± ↑ granulocytes and platelets in the absence of physiologic stimulus
Etiologies of erythrocytosis (absolute ↑ RBC)
• Acquired 1°: PV (usually JAK2+) or other MPN
• Germline 1°: Chuvash (hypoxia-sensing disorder due to VHL mutation), EGLN mutations
• Secondary: carboxyhemoglobinemia; hypoxia (OSA, COPD); inappropriate erythropoietin (renal, hepatic); Cushing’s syndrome
• Mimics: relative ↑ RBC (↓ plasma) due to dehydration; “stress” erythrocytosis (Gaisböck’s syndrome)
Clinical manifestations (common between PV and ET)
• Symptoms (often termed “vasomotor”)
Hyperviscosity (erythrocytosis): headache, dizziness, tinnitus, blurred vision
Thrombosis (hyperviscosity, thrombocytosis): ↑ risk of DVT, MI, stroke; transient visual disturbances (amaurosis, ocular migraine); Budd-Chiari syndrome; erythromelalgia = intense burning, pain and erythema of extremities due to microvascular ischemia
Bleeding (abnormal platelet function): easy bruising, epistaxis, GI bleeding
↑ histamine from basophils → pruritus, peptic ulcers; ↑ uric acid (cell turnover) → gout
• Signs: plethora, splenomegaly, hypertension, engorged retinal veins
Diagnostic evaluation
• Men: Hb >16.5 g/dL or Hct >49%, women: Hb >16 g/dL or Hct >48%
• ± ↑ WBC, platelets, basophils; ↑ uric acid, leukocyte alkaline phosphatase, vit B12
• Peripheral smear → no morphologic abnormalities
• ✓ Epo to rule out secondary causes of erythrocytosis; if Epo ↓, PV more likely If Epo ↑, then ✓ SaO2 or PaO2, carboxyhemoglobin, BM exam
• BM bx → hypercellularity for age, trilineage growth, pleomorphic mature megakaryocytes
• JAK2 V617F mutation in ~95% of PV; other Pts typically harbor JAK2 exon 12 mutations
Treatment for JAK2 + PV
• Phlebotomy to goal Hct <45% (NEJM 2013;368:22), consider <42% in women
• Low-dose ASA in all Pts (NEJM 2004;350:114)
• Hydroxyurea if high risk of thrombosis (age ≥60, prior thrombosis) or if inadequate Hct by phlebotomy alone
• PEG IFNα preferred in younger Pts and pregnancy (Lancet Haematol 2017;4:e165)
• Ruxolitinib (JAK1/2 inhibitor) if refractory to or intolerant of hydroxyurea (NEJM 2015;372:426)
• Supportive: allopurinol (gout), H2-blockers/antihistamines (pruritus). Avoid iron supp.
• Data for optimal mgmt of other types of PV (secondary, germline, etc.) currently lacking
Prognosis
• Median survival w/ Rx ~13.5 y (Blood 2014;124:2507); ↑ age, WBC, additional acquired somatic mutations → worse prognosis (Haematol 2013;160:251)
• Post-PV myelofibrosis (spent phase) occurs in 10–20% of cases, usually after 10 y
• Risk of transformation into acute leukemia (<2–5% lifetime)
ESSENTIAL THROMBOCYTHEMIA (ET)
Definition
• Sustained ↑ in platelets (>450,000/µL) ± ↑ RBC and granulocytes
Etiologies of thrombocytosis
• 1° = ET or other MPN; myelodysplastic syndromes (5q-syndrome); RARS-T
• 2° = reactive thrombocytosis: inflammation (RA, IBD, vasculitis), infection, trauma, acute bleed, iron deficiency, postsplenectomy, neoplasms (eg, Hodgkin lymphoma)
• Of patients with plt >1,000,000/µL, <1 in 6 will have ET
Clinical manifestations (also see “Polycythemia Vera”)
• Thrombosis with erythromelalgia (risk of thrombosis highest in Pts with leukocytosis), bleeding (acquired vWD), pruritus; mild splenomegaly; migraine, TIA; early fetal loss
Diagnostic evaluation
• Peripheral smear: large hypogranular platelets
• BM bx: megakaryocytic hyperplasia; ⊖ Phil chr; rarely minor reticulin fibrosis; nl Fe; if atypical megakaryoctyes or ↑ reticulin, consider pre-PMF (Rx same as ET, but ↑ risk MF)
• Mutations: JAK2 V617F in ~50%; CALR in ~45%; MPL in 5–10%; triple negative <5%
• Patients should not meet WHO criteria for diagnosis of CML, PV, PMF, or MDS
• Check vWF in pts w/ plt>1,000,000 and hold ASA (vide infra) if acquired vWD
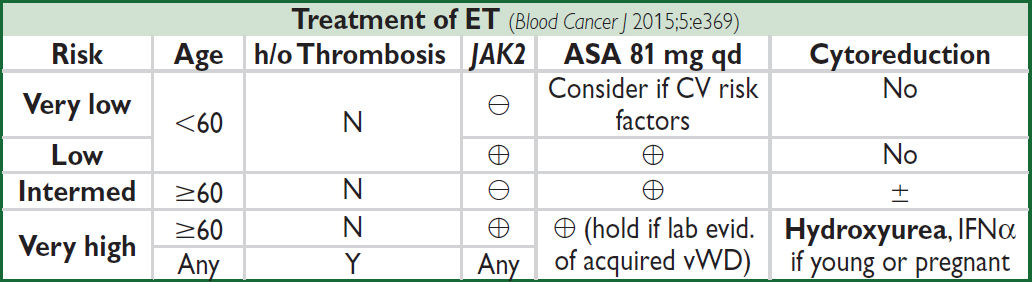
Prognosis
• Low-risk Pts have overall survival ≈ control population
• Risk of transformation into acute leukemia <2–3% lifetime; risk of progression to MF ~10%
PRIMARY MYELOFIBROSIS (PMF)
Definition
• Clonal myeloproliferation with reactive marrow fibrosis & extramedullary hematopoiesis
• Prefibrotic stage (pre-PMF): megakaryocyte prolif, grade 1 reticulin fibrosis, ↑ BM cellularity. Compared w/ ET, pre-PMF has ↑ thrombosis, ↑ progression, ↓ survival (Blood 2012;120:569).
Etiologies of myelofibrosis
• 1° myelofibrosis (PMF): myeloproliferative neoplasm
• 2° myelofibrosis: post-PV/ET myelofibrosis, other hematologic (CML, AML, ALL, MDS) and solid cancers (breast, prostate), autoimmune (eg, SLE), toxin (benzene), radiation, granulomas (TB, fungal, sarcoid), deposition diseases (eg, Gaucher’s)
Clinical manifestations (BJH 2012;158:453)
• Ineffective erythropoiesis → anemia; extramedullary hematopoiesis → massive splenomegaly (abdominal pain, early satiety) ± hepatomegaly
• Tumor bulk and ↑ cell turnover → fatigue, weight loss, fever, sweats
Diagnostic evaluation (Blood 2010;115:1703 & 2016;127:2391)
• Anemia with variable WBC and platelet counts
• Peripheral smear → “leukoerythroblastic” (teardrop cells, nucleated RBCs, immature WBCs); large abnormal platelets
• BM aspirate → “dry” tap; BM bx → severe fibrosis, replacement by reticulin & collagen
• JAK2 V617F in 45–50%; CALR mut in 45–50%, MPL mut in 7–10%, triple neg in 1–2%
• No BCR-ABL translocation; Pts should not meet criteria for PV or MDS
• DIPSS score for prognosis. High-risk factors: age >65, WBC >25k, Hgb <10, peripheral blasts >1%, symptoms, complex cytogenetics, absence of CALR type 1.
Treatment (Am J Hematol 2021;96:145)
• In absence of adverse prognostic factors (eg, anemia or sx) → no treatment
• Allogeneic HSCT only potential cure → consider in young Pts w/ high-risk disease
• Supportive care: transfusions; ESA if Epo <500 (risk ↑ splenomegaly); consider androgens vs. immunomodulatory agents (eg, lenalidomide) + prednisone; hydroxyurea; ? splenectomy if refractory to transfusions, failed chemoRx, painful splenomegaly
• JAK inh: ruxolitinib (JAK1/2) ↓ sx, ↓ splenomegaly, ↑ survival; preferred (NEJM 2012;366:787 & 799); fedratinib (JAK2; JAMA Oncol 2015;1:643); pacritinib & momelotinib under study
• Median survival ~6 y (JCO 2012;30:2981); transformation into AML occurs at a rate of ~8%/y
LEUKEMIA
ACUTE LEUKEMIA
Definition
• Clonal proliferation of hematopoietic progenitor with failed differentiation into mature elements → ↑ blasts in bone marrow and periphery → ↓ RBCs, platelets, and neutrophils
Epidemiology and risk factors
• Acute myelogenous (AML): ~20k cases/y in U.S.; median age 68 y
• Acute lymphocytic (ALL): ~6k cases/y in U.S.; median age 15 y but 2nd peak in older adults
• Risk factors: radiation, chemo (alkylating agents, topo II inhib), benzene, smoking, ? rising from acquired somatic mutations and clonal hematopoiesis (NEJM 2014;371:2477)
• Secondary to acquired hematopoietic dis.: MDS, MPN (esp. CML), aplastic anemia, PNH
• Inherited: Down’s, Klinefelter’s, Fanconi’s anemia, Bloom synd., ataxia telangiectasia, germline mut. in TP53 (Li-Fraumeni syndrome), DDX41, RUNX1, CEBPa, & GATA2
Clinical manifestations
• Cytopenias → fatigue (anemia), infection (neutropenia), bleeding (thrombocytopenia)
• Leukostasis (more in AML): blast >50k, ↓ SaO2, HA, blurry vision, confusion, TIA/CVA
• Tumor lysis syndrome (TLS) from rapid turnover of cells
• Disseminated intravascular coagulation (DIC; especially with APL)
• Other: leukemic infiltration of skin, gingiva (esp. with monocytic subtypes); chloroma (myeloid sarcoma): extramedullary tumor of leukemic cells, any location; anterior mediastinal mass and SVC syndrome (T-ALL/LBL); hepatosplenomegaly (ALL and monocytic leukemias); CNS (~10% of ALL; also in monocytic >myeloid leukemias): cranial neuropathies, HA
Diagnostic evaluation (Blood 2009;114:937)
• Peripheral smear: thrombocytopenia, blasts (seen in >95%; ⊕ Auer Rods in AML)
• Bone marrow: >20% blasts; mostly hypercellular; test for cytogenetics and flow cytometry for immunophenotype (AML/ALL)
• Cytogenetic anomalies: eg, in AML, t(15;17), t(8;21), inv(16) or t(16;16), complex; in ALL, Ph-chromosome [t(9;22)], hyper or hypodiploid, complex
• Molecular mutations in AML: esp FLT3 (ITD and TKD), TP53, NPM1; ALL: BCR-ABL1
• Evaluate for complications: TLS (↑ uric acid, ↑ LDH, ↑ K, ↑ PO4, ↓ Ca), DIC (PT, PTT, fibrinogen, D-dimer, haptoglobin, bilirubin), check for G6PD (prior to giving rasburicase)
• LP (w/ co-admin of intrathecal chemotherapy to avoid seeding CSF w/ circulating blasts) for all Pts w/ ALL (CNS is sanctuary site) and for Pts w/ AML w/ CNS sx
• TTE before use of anthracyclines
• HLA typing of Pt, siblings >parents/children for potential allogeneic HSCT candidates
ACUTE MYELOGENOUS LEUKEMIA (AML)
Classification (WHO; Blood 2016;127:2391)
• Features used to confirm myeloid lineage and subclassify AML to guide treatment: morphology: blasts, ⊕ granules, ± Auer rods (eosinophilic needle-like inclusions)
• Immunophenotype: precursor: CD34, CD45, HLA-DR; myeloid: CD13, CD33, CD117; monocyte: CD11b, CD64, CD14, CD15
• Histochem.: myeloperoxidase (myeloid), non-specific esterase, and lysozyme (monocytic)
• Prognosis: age, prior antecedent MPN/MDS and genetics (cytogenetics + molecular mutation status) are key independent risk factors of poor prognosis
ENL 2017 Genetic Risk Classification (Blood 2017;129:424) |
|
Risk Category |
Genetic Abnormality |
Favorable |
APL: t(15;17); PML-RARa; t(8;21): RUNX1-RUNX1T1; inv(16): CBFB-MYH1; mutated NPM1 w/o FLT3-ITD or w/ FLT3-ITDlow*; biallelic mutation in CEBPA |
Intermediate |
FLT3-ITDlow*; mutated NPM1 & FLT3-ITDhigh*; t(9;11): MLL-MLLT3; cytogenetic abnl not classified as favorable or adverse, including normal karyotype w/o mutations in FLT3-ITD & NPM1 |
Adverse |
-5 or del(5q); -7; -17/abn(17p); complex or monosomal karyotype; t(6;9): DEK-NUP214; t(9;22) BCR-ABL1; inv(3): GATA2-MECOM; wildtype NPM1 & FLT-ITDhigh*; mutated TP53, RUNX1, ASXL1 |
* low/high: FLT3-ITD variant allele frequency (VAF); reflects burden of mut. leukemic cells |
|
Upfront treatment
• Induction chemo “7+3”: 7d cont. infusion cytarabine (Ara-C) + 3d bolus anthracycline (daunorubicin or idarubicin)
• Ability to tolerate 7+3 regimen key determinant in subsequent Rx received (vide infra)
• Obtain BM bx 14–21 days after start of induction chemo to assess response
• Regimens for fit (generally age <75 y)
FLT3-ITD/TKD mutation: 7+3+midostaurin (early generation FLT3 inhib; NEJM 2017;377:454)
Core-binding factor ⊕: t(8;21) or inv(16): 7+3 ± gemtuzumab ozogamicin (mAb targ. CD33)
2° AML or w/ MDS-related changes: CPX-351 (liposomal Ara-C & daunorubicin)
Other: age <60 y: 7+3 (high-dose daunorubicin 90 mg/m2); >60 y: dauno 60 mg/m2
• Regimens for unfit (may include age ≥75 y or < 75y w/ ECOG ≥3 or severe cardiac or pulmonary comorbidity; Leukemia 2013;27:997)
Azacitadine + venetoclax (Bcl2 inhibitor) (NEJM 2020;383:617)
IDH1/2 mutation: ivosidenib or enasedinib
Consolidation therapy
• If complete remission (CR) = ANC >103, plt >100, no RBC Rx, <5% BM blasts; CR ≠ cure
• Favorable risk: high-dose cytarabine (HiDAC); Intermediate/Poor risk: Allo-HSCT
• Consider maintenance azacitadine if cannot complete curative intent Rx (NEJM 2020;383:2526)
Refractory/relapsed disease
• Repeating mutation analysis key b/c clonal evolution common and may affect Rx
• FLT3-ITD/TKD mutation: gilteritinib (potent FLT3 inhibitor)
• IDH1 mutation: ivosidenib; IDH2 mutation: enasidenib (small-molecule inhib of IDH1 or 2)
• Chemo: MEC (mitoxantrone, etoposide, Ara-C); FLAG-Ida (fludarabine, Ara-C, G-CSF, & idarubicin); CLAM (clofarabine, Ara-C, mitoxantrone), gemtuzumab
Prognosis
• CR achieved in 70–80% of Pts <60 y and in 40–50% of Pts >60 y
• Overall survival variable, depends on prognostic factors: ranges from <10% of older Pts w/ poor-risk tumor genetics to >65% of younger Pts w/ favorable prognostic factors
Acute promyelocytic leukemia (APL) (Blood 2009;113:1875)
• Rare, ~8% of AML in U.S.; >90% cure rates
• Atypical promyelocytes (large, granular cells; bilobed nuclei) in blood and bone marrow
• Defined by translocation of retinoic acid receptor: t(15;17); PML-RARA (>95% of cases)
• Medical emergency with DIC and bleeding common
• Remarkable responses to all-trans-retinoic acid (ATRA) & arsenic trioxide (ATO) which induce differentiation of leukemic blasts. ∴ early initiation of ATRA if APL suspected
• Non-high-risk APL: ATRA + ATO (induction + 4 cycles consolidation) → CR ~100%; event-free survival 97% and overall survival 99% at 2 y (NEJM 2013;362:111)
• High-risk APL: WBC >10k at diagnosis. No clear consensus. In general, chemo (anthracycline or gemtuzumab ozogamicin) added to ATRA + ATO induction and consolidation.
• Differentiation syndrome (ATRA): ~25% of Pts; fever, pulm. infiltrates, SOB, edema, HoTN, AKI; tx w/ dexamethasone 10 mg bid, supportive care (eg, diuresis) (Blood 2008;113:775)
ACUTE LYMPHOBLASTIC LEUKEMIA (ALL; Lancet 2020;395:1146)
Classification
• Lymphoblastic neoplasms may present as acute leukemia (ALL) w/ >20% BM blasts or as lymphoblastic lymphoma (LBL, more common in T-cell) w/ mass lesion w/ <20% BM blast
• Morphology: no granules (granules seen in myeloid lineage)
• Cytochemistry: ⊕ terminal deoxynucleotidyl transferase (TdT) in 95% of ALL, MPO ⊖
• Immunophenotype
Precursor: CD34, TdT
B: CD19; variable CD10, CD22, CD79a
T: CD1a, CD2, cytoplasmic CD3, CD5, CD7
Treatment
• Induction chemo
Ph ⊕ t(9;22) (seen in ~25% of B-ALL): tyrosine kinase inhibitor + chemo/steroids
Ph ⊖: Adolescents & young adults (<40 y): pedi-like regimen typically w/ PEG-asparaginase. Adults (40–60 y): multiagent chemo incl. anthracycline, vincristine, steroids, cyclophosphamide (CYC). Older (>60 y): reduced-intensity chemo.
• CNS prophylaxis: intrathecal MTX/cytarabine ± cranial irradiation or systemic MTX
• Post-remission therapy (choice depends on risk of recurrence)
1) Average risk: consolidation/intensification chemo (~7 mo) → maintenance (~2–3 y)
2) High risk: high-dose chemo w/ allo-HSCT considered for Pts in CR1. High-risk disease includes: Ph ⊕; Ph-like (based on gene expression); MLL translocation t(4;11); complex karyotype; hypodiploid (<44 chromosomes); early T-cell phenotype (ETP; lacks CD1a, CD8, CD5weak, myeloid markers); minimal residual disease (MRD) = morphologic remission but flow cytometry or molec. markers of tumor still detectable.
• Relapse/refractory: salvage therapy (below), then allogeneic HSCT if able
B cell: blinatumomab (CD19 BiTE-bispecific T-cell engager; NEJM 2017;376:836), inotuzumab (CD22 Ab drug conjugate; NEJM 2016;375:740); tisagenlecleucel and brexucabtagene autoleucel (CD19 CAR-T, NEJM 2018;378:449; Lancet 2021;398:491), TKI+chemo/steroids (Ph ⊕t(9;22) only)
T cell: nelarabine ± cyclophosphamide and etoposide
Both B & T cell: chemo including high dose cytarabine regimens; clofarabine
CHRONIC MYELOGENOUS LEUKEMIA (CML; Lancet 2021;398:1914)
Definition (Blood 2009;114:937)
• Myeloproliferative neoplasm with clonal overproduction of hematopoietic myeloid stem cells that can differentiate
• Philadelphia chromosome (Ph) = t(9;22) → BCR-ABL1 fusion → ↑ Abl kinase activity
BCR-ABL1 required for diagnosis (make via karyotyping or FISH; PCR)
Epidemiology and risk factors
• ~6600 new cases/y in U.S.; median age ~64 at presentation; ~15% of adult leukemias
• ↑ risk with irradiation; no clear relation to cytotoxic drugs
Disease classification & manifestations (WHO; NCCN v 2.2022)
• Chronic phase (CP): <10% blasts (peripheral or bone marrow). Risk stratification based on Sokal (Blood 1984;63:789) or Euro scores (J Clin Pathol 2001;54:491).
• Accelerated phase (AP): 10–19% blasts, ≥20% basos, plts <100k, clonal evolution (karyotype changes) not seen at dx
• Blastic phase (BP): ≥20% blasts (peripheral or marrow) and/or extramedullary leukemia
• Most Pts asx or may have mild constitutional s/s related to splenomegaly.
• Worsening constitutional sx, bone pain, rapid ↑ in spleen size herald disease progression
Diagnostic evaluation
• Peripheral smear: leukocytosis, left-shifted with all stages of myeloid maturation; thrombocytosis, basophilia
• Bone marrow w/ karyotype: hypercellular, ↑myeloid:erythroid ratio, micromegakaryocytes
Treatment (Lancet 2015;385:1447; Hematol Oncol Clin North Am 2017;31:577)
• Tyrosine kinase inhibitors (TKI) inhibit abl kinase activity.
1st gen: imatinib, 1st TKI against BCR-ABL1 (NEJM 2017;376:917)
2nd gen: nilotinib, dasatinib, bosutinib. ↑ response but ↑ toxicity. No survival difference.
3rd gen: ponatinib; a/w ↓ risk of disease progress, preferred for int-high risk, but ↑ toxicity
Imatinib, dasatinib, nilotinib, & bosutinib approved for 1st line Rx. Nilotinib, dasatinib, bosutinib, ponatinib, & asciminib approved for resistant disease; only ponatinib & asciminib effective on T315I mutation (NEJM 2012;367:2075, Blood 2021;138:2031).
STAMP (allosteric inhibitor): asciminib (NEJM 2019;381:2315); after ≥2 prior TKIs
Resistance: due to ↑ BCR-ABL1 expression, often 2/2 ABL kinase mutation or amplification
Side effects: N/V, diarrhea, muscle cramps, cytopenias, ↓ PO4, ↑ QT, rarely CHF; dasatinib: pericardial & pleural effusions and pulm. HTN; nilotinib: ↑ bili & lipase, CV toxicity; ponatinib: pancreatitis and arterial vascular events (cerebral, cardiac, & PAD)
• TKI discontinuation: consider if complete molecular response (>4.5 log reduction in BCR-ABL1 transcript) for >2 y. Up to 50% of Pts remain off TKI at 2 y (ie, no molec. recurrence). Success proportional to duration of CMR and risk score at presentation.
• Consider allogeneic HSCT for AP and BP.
• CML in pregnancy: hydroxyurea & all TKIs contraind. If Rx needed, IFN (only option).
Milestones of Therapy |
|
Definition |
Optimal Time |
BCR-ABL1 ratio <10% IS = 1-log reduction by QT- PCR |
3 mo |
BCR-ABL1 ratio <1% IS = 2-log reduction by QT-PCR (CCyR) |
6 mo |
BCR-ABL1 ratio <0.1% IS = 3-log reduction by QT-PCR (MMR) |
12 mo |
BCR-ABL1 ratio: Pt BCR-ABL1 mRNA: ABL mRNA in peripheral blood. International Scale (IS) standardizes across labs by normalizing to BCR-ABL1:ABL1 ratio in a cohort of unRx’d patients. |
|
Prognosis (NEJM 2017;376:917)
• Chronic phase CML Rx’d w/ imatinib: 89% 5-y overall survival, 95% survival free of CML-related deaths, 7% progression to blast phase at 5 y (NEJM 2006;355:2408). Pts in CCyR (~equal to QT-PCR if 1% IS) have normal life expectancy. QT-PCR >4-log ↓ can consider TKI discontinuation trial (Lancet Onc 2018;19:747).
CHRONIC LYMPHOCYTIC LEUKEMIA (CLL)
see “Small Lymphocytic Lymphoma”
LYMPHOMA AND CLL
Definition
• Malignant disorder of lymphoid cells that reside predominantly in lymphoid tissues
• Generally characterized as Hodgkin lymphoma (HL) or non-Hodgkin lymphoma (NHL)
Clinical manifestations
• Lymphadenopathy (usually nontender)
HL: superficial (usually cervical/supraclavicular) ± mediastinal LAN; nodal disease with orderly, anatomic spread to adjacent nodes
NHL: diffuse; nodal and/or extranodal disease with noncontiguous spread; symptoms reflect involved sites (abdominal fullness, bone pain)
• Constitutional (“B”) symptoms: fever (>38°), drenching sweats, ↓ weight (>10% in 6 mo)
HL: periodic, recurrent “Pel-Ebstein” fever; 10–15% have pruritus; ~35% “B” symptoms
NHL: “B” symptoms vary between subtypes, ~15–50%
Diagnostic and staging evaluation
• Physical exam: lymph nodes, liver/spleen size, Waldeyer’s ring, testes (~1% of NHL), skin
• Pathology: excisional lymph node bx (not FNA b/c need surrounding architecture) with immunophenotyping and cytogenetics (Reed-Sternberg (RS) cells in HL); BM bx if cytopenias; CLL by peripheral flow in patients w/ peripheral disease; LP if CNS involvement clinically suspected
• Lab tests: CBC, BUN/Cr, LFTs, ESR, LDH, UA, Ca, alb; ✓ HBV & HCV (and must ✓ HBsAg & anti-HBc if planning rituximab Rx due to risk of HBV reactivation); HIV
• Imaging: PET-CT to assess disease burden and guide biopsy/therapy, especially in HL, DLBCL. Head CT/MRI only if neurologic symptoms.
Ann Arbor Staging System with Cotswolds Modifications |
|
Stage |
Features |
I |
Single lymph node (LN) region |
II |
≥2 LN regions on the same side of the diaphragm |
III |
LN regions on both sides of the diaphragm |
IV |
Disseminated involvement of one or more extralymphatic organs |
Modifiers: A = no symptoms; B = fever, night sweats or weight loss; X = ”bulky” mediastinal disease or mass >10 cm; E = single contiguous extranodal site; H = hepatic; S = splenic |
|
HODGKIN LYMPHOMA (HL) (Am J Hematol 2018;93:704; Lancet 2021;398:1518)
Epidemiology and risk factors
• ~9,000 cases/y; bimodal distribution (15–35 & >50 y); ↑ ♂; role of EBV in subsets of HL, esp. immunocompromised patients (eg, HIV)
Pathology
• Affected nodes show RS cells (<1%) in background of non-neoplastic inflammatory cells
• Classic RS cells: bilobed nucleus & prominent nucleoli with surrounding clear space (“owl’s eyes”). RS cells are clonal B-cells: CD15+, CD30+, CD20– (rarely +).
WHO Histologic Classification of Classical HL |
||
Nodular sclerosis |
60–80% |
Collagen bands; frequent mediastinal LAN; young adults; female predominance; usually stage I or II at dx |
Mixed cellularity |
15–30% |
Pleomorphic; older age; male predominance; ≥50% stage III or IV at presentation; intermediate prognosis |
Lymphocyte rich |
5% |
Abundant normal-appearing lymphocytes; mediastinal LAN uncommon; male predominance; good prognosis |
Lymphocyte depleted |
<1% |
Diffuse fibrosis and large numbers of RS cells; older, male patients; disseminated at dx; seen in HIV; worst prognosis |
• Nonclassical HL (5%): nodular lymphocyte predominant (NLP); involves peripheral LN
80% present in stages I–II, Rx w/ RT vs. chemoRT w/ 4-yr PFS 88% and OS 96% (JCO 2008;26:434); consider rituximab because most NLP RS cells are CD20+
Stages III–IV treated with combination chemo (see below)
Treatment (NCCN Guidelines v5.2021)
• Stages I–II: PET adapted ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine) ± RT Favorable: ABVD × 2–4 cycles ± RT (if PET ⊖ after 2 cycles reduce chemo or RT)
Unfavorable (>50 y, bulky mediastinum, “B” sx, ESR >50, >3 nodal sites): ABVD × 4–6 + RT (stop at 4 cycles if PET ⊖ after 2 cycles)
• Stages III–IV: ABVD × 6 cycles (can omit B if PET ⊖ after 2 cycles; NEJM 2016;374:2419); brentuximab (anti-CD30) may replace bleo but more toxic (NEJM 2018;378:331); add RT for select Pts as consolidation
• Refractory/relapsed disease: salvage chemo + auto HSCT ± RT
brentuximab vedotin post-ASCT yields some long-term remissions (Blood 2016;128:1562)
PD1/PDL1 blockade (pembrolizumab or nivolumab) (NEJM 2015;372:311, JCO 2017;35:19)
• Late treatment effects include ↑ risk for:
Second cancers: ~4.6× risk for up to 40 y (NEJM 2015;373:2499)
breast (if RT), ∴ annual screening at age 40 or 8–10 y post RT; leukemia/MDS; NHL
Cardiac disease (if RT or anthracycline), ? role of echo/stress at 10 y (controversial)
Pulmonary toxicity (if bleomycin); Hypothyroidism (if RT), ∴ annual TSH (if neck RT)
• International Prognostic Score (IPS; JCO 2012;30:3383) risk factors: albumin <4 g/dL, Hb <10.5 g/dL, male gender, age >45 y, stage IV, WBC ≥15 k/µL, lymphocytes <600/µL or <8% of diff. 5-yr PFS 62–88% based on # of risk factors.
NON-HODGKIN LYMPHOMA (NHL)
Epidemiology and risk factors
• 79 types of NHL, ~70,000 new cases/y; median age dx 65 y; ♂ predom; 85% B-cell origin
• Associated conditions: immunodeficiency (eg, HIV, posttransplant); autoimmune
disorders (eg, Sjögren’s, RA, SLE); infection (eg, EBV, HTLV-I, H. pylori)
• Burkitt lymphoma: (1) endemic or African (jaw mass, 80–90% EBV-related); (2) sporadic or American (20% EBV-related); (3) HIV-related
WHO Classification of Lymphoid Malignancies (Blood 2016;127:2375) |
||
Type |
Examples |
Associated Abnormalities |
Mature B cell
aggressiveness |
Burkitt’s lymphoma Diffuse large B-cell lymphoma (DLBCL) Mantle cell Marginal zone lymphoma (nodal, extranodal, splenic) Hairy cell leukemia (⊕ TRAP) Follicular lymphoma CLL/small lymphocytic lymphoma |
8q24, c-MYC BCL2, MYC, MLL2, CREBBP, etc. t(11;14) BCL1-IgH → cyclin D1 AP12-MALT1 & BCL-10-Ig enh
BRAF V600E IGH-BCL2, MLL2, CREBBP IGHV, TP53, ATM, SF3B1, etc. |
Mature T cell & NK cell |
Peripheral T-cell lymphoma Mycosis fungoides (cutaneous lymphoma)/ Sézary syndrome (+ LAN) Anaplastic large-cell lymphoma Angioimmunoblastic T-cell lymphoma |
TET2 and DNMT3A
Some ALK1 ⊕ |

Treatment (Lancet 2017;390:298)
• Treatment and prognosis determined by histopathologic classification rather than stage
• Rituximab (anti-CD20; NEJM 2012;366:2008) if CD20+
• Indolent: generally no cure (except allo HSCT), goal sx mgmt (bulky dis, cytopenia, “B” sx)
Follicular Lymphoma Int’l Prog. Index (FLIPI; Blood 2004;104:1258): risk factors = age >60, stage III/IV, Hb <12 g/dL, >4 nodal areas, LDH >nl. 5-yr OS 52–90% based on score.
Initial: RT if localized, rituximab + chemo (bendamustine, CVP, fludarabine), ibrutinib.
Obinutuzumab (anti-CD20) + chemo w/ obin maint ↑ PFS but ↑ tox (NEJM 2017;377:1331).
Maintenance: rituximab in indolent, aggressive, and relapsed disease (Lancet 2011;377:42)
Hairy cell: cladribine; oral BRAF inhibitor if relapsed/refractory (NEJM 2015;373:1733)
Gastric MALT: ✓ H. pylori; can cure by treating H. pylori if ⊕; RT for relapsed/refractory
• Aggressive: goal is cure (Am J Hematol 2019;94:604), treatment depends on subtype
International Prognostic Index (IPI; Blood 2007;109:1857): risk factors = age >60 y, stage III/IV, ≥2 extranodal sites, PS ≥2, LDH >nl. 4-yr OS 55–94% based on score.
R-CHOP (rituximab, cyclophosphamide, doxorubicin = hydroxydaunorubicin, vincristine = Oncovin, prednisone) (NEJM 2002;346:235 & 2008;359:613) DLBCL 10-y PFS = 45%; overall survival = 55% (Blood 2010;116:2040)
+ Radiation for localized or bulky disease
Consider CNS prophylaxis w/ intrathecal vs. systemic high-dose MTX if paranasal sinus, testicular, breast, periorbital, paravertebral, or bone marrow involved; also w/ ≥2 extranodal sites + ↑ LDH. Controversial (Blood 2021;139:413).
Refractory/relapsed disease: salvage chemo; high-dose chemo + auto-HSCT (JCO 2001;19:406); allo-HSCT if beyond 2nd relapse (JCO 2011;29:1342)
CAR-T (qv): axicabtagene (NEJM 2017;377:2531), tisagenlecleucel (NEJM 2019;380:45), lisocabtagene (Lancet 2020;396:839), brexucabtagene (mantle cell; NEJM 2020; 382:1331)
Mantle cell: ibrutinib for relapsed/refractory disease (Lancet 2016;387:770)
Burkitt: dose-adjusted EPOCH-R (NEJM 2013;369:1915) or CODOX-M/IVAC (cyclophosphamide, vincristine, doxorubicin, high-dose methotrexate, ifosfamide, etoposide, high-dose cytarabine rituximab) (Blood 2008;112:2248)
All Pts receive CNS & tumor lysis syndrome prophylaxis
Rituximab ↑ event-free survival (Lancet 2016;387:2402)
High-grade B-cell lymphoma w/ rearrangements of MYC and BCL2 and/or BCL6: “double-/triple-hit”, assoc. w/ poor prognosis. Often use DA-R-EPOCH.
HIV-associated NHL (Blood 2006;107:13)
• HIV ⊕ imparts 60–100× relative risk
• NHL is an AIDS-defining malignancy along with Kaposi’s, cervical CA, anal CA
• Concurrent HAART & chemotherapy likely provide survival benefit
• DLBCL & immunoblastic lymphoma (67%): CD4 <100, EBV-associated. Burkitt lymphoma (20%): can occur with CD4 >200. Generally, treat as immunocompetent, but avoid rituximab if CD4 <50 (due to increased risk of death from infection).
• Primary CNS lymphoma (16%): CD4 <50, EBV-assoc. (also seen in Pts w/o HIV). Rx w/ high-dose MTX-based regimen + steroid ± temozolomide ± RT, consider auto HSCT.
SMALL LYMPHOCYTIC LYMPHOMA (SLL) OR CHRONIC LYMPHOCYTIC LEUKEMIA (CLL)
Definition (NEJM 2005;352:804; Blood 2008;111:5446)
• Monoclonal, functionally incompetent mature B lymphocytes; CLL & SLL = same disease
• CLL: >5000/µL malignant cells; SLL: <5000/µL malignant cells + LAN ± splenomegaly
• Monoclonal B lymphocytosis: resembles but does not meet CLL criteria, observe
Epidemiology and risk factors
• ~15,000 new cases/y; median age at dx is 71 y; most common adult leukemia
Clinical manifestations
• Often asx & identified by lymphocytosis on CBC; 10–20% p/w “B symptoms”
• Lymphadenopathy (80%) and hepatosplenomegaly (50%)
• Autoimmune hemolytic anemia (AIHA) (~10%) or thrombocytopenia (ITP) (~1–2%)
• Hypogammaglobulinemia ± neutropenia → ↑ susceptibility to infections
• Bone marrow failure in ~13%; monoclonal gammopathy in ~5%
• Aggressive Richter’s transformation in ~5% into high-grade lymphoma (poor prognosis)
Diagnostic evaluation
• CBC w/ diff: B-cell count
• Peripheral smear: lymphocytosis (predominantly mature-appearing small cells) “smudge” cells from damage to abnl lymphs from shear stress of making blood smear
• Flow cytometry: clonality with dim surface Ig; CD5+, CD19+, CD20+, CD23+
• Bone marrow (not req for dx): normo- or hypercellular; ≥30% small B lymphocytes
• Genetics: del 11q22-23 & del(17p) unfavorable; trisomy 12 neutral; del 13q14 and mut IGHV favorable. Nine significantly mutated genes, including TP53, NOTCH1, MYD88, and SF3B1. Key role for spliceosome mutations (NEJM 2011;365:2497; JCI 2012;122:3432).
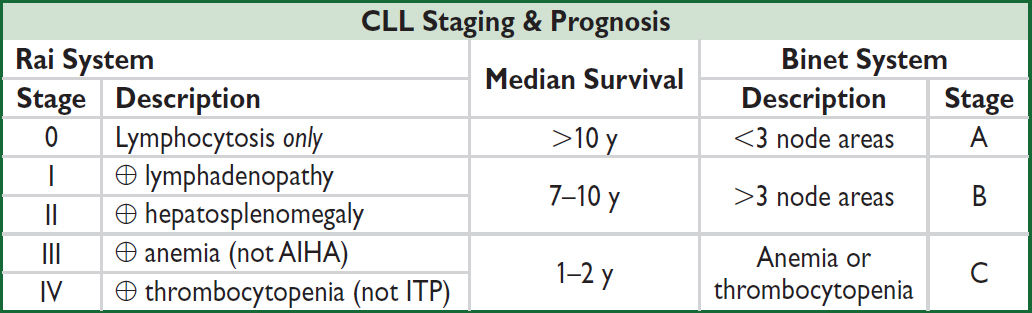
Treatment (NEJM 2020;383:460)
• Observation unless “active disease”: Rai stage III/IV, Binet stage C, disease-related sx, progressive disease, AIHA or ITP refractory to steroids, recurrent infections
• First-line: w/o del(17p)/TP53 use acalabrutinib or ibrutinib (BTK inhibitors) or venetoclax + rituximab; with del(17p)/TP53 use acalabrutinib or venetoclax ± obinutuzumab; ibrutinib + venetoclax w 88% w/ CR but ↑ tox (NEJM 2019;380:2095)
• Second-line & beyond: in general, choose Rx w/ mechanism different from 1st line Rx. BTK inhibitors (eg, zanubrutinib), venetoclax, chemo including fludarabine, chlorambucil, or bendamustine + rituximab. Consider allo-HSCT in relapse.
• HSCT is the only curative Rx. Rx choice balances patient/disease characteristics and goals of care. Different rates of complete remission, time to progression, and toxicities.
• Rx for complications: PCP, HSV, VZV Ppx; AIHA/ITP → steroids; recurrent infxns → IVIg
PLASMA CELL DYSCRASIAS
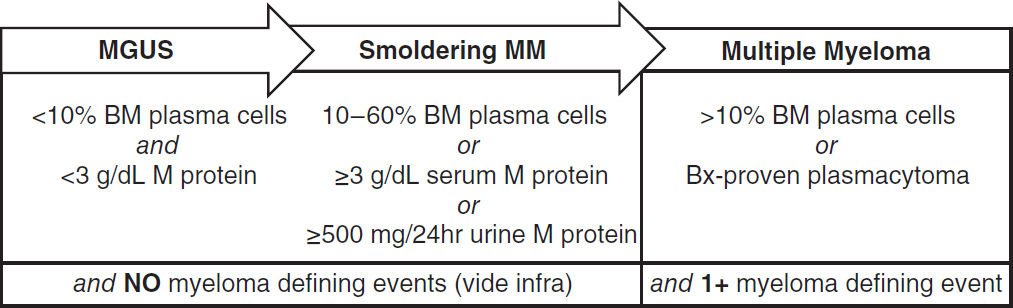
Monoclonal Gammopathy of Uncertain Significance (MGUS) (NEJM 2018;378:241)
• ✓ CBC, Ca, Cr, SPEP, serum free light chains, UPEP w/ immunofixation (to exclude MM)
• Close observation: repeat SPEP in 6 mo, then yearly thereafter if stable
• ~1%/y or ~25% lifetime risk → MM, WM, amyloidosis, or malign. lymphoproliferative dis.
• Abnormal serum free light chain ratio & M protein ≥1.5 g/dL: ↑ risk of progression to MM
Smoldering Multiple Myeloma
• Need whole-body MRI or PET-CT to rule out occult bone lesions
• Risk of prog. 10%/y, depends on [M protein], subtype, FLC ratio. No defined role for Rx yet though trials ongoing (NEJM 2013;369:438; Blood 2019;134:781; JCO 2020;38:1126).
MULTIPLE MYELOMA (MM)
Definition and epidemiology (Lancet 2021;397:410)
• Malignant neoplasm of plasma cells producing a monoclonal Ig = “M protein”
• ~27,000 new cases/y; median age at diagnosis 69 y; more common in African Americans
Clinical manifestations (CRAB criteria and other less common features)
• HyperCalcemia due to ↑ osteoclast activity
• Renal disease: multiple mechanisms include toxic effect of filtered light chains → renal failure (cast nephropathy) or type II RTA; amyloidosis or light chain deposition disease → nephrotic syndrome; hypercalcemia, urate nephropathy, type I cryoglobulinemia
• Anemia (normocytic) due to bone marrow involvement; rarely, may see AIHA
• Lytic Bone lesions due to ↑ osteoclast activity → pathologic fx
• Recurrent infxns due to relative hypogammaglob. (clonal plasma cells suppress nl Ig)
• Neurologic: cord compression; POEMS (polyneuropathy, organomegaly, endocrinopathy, M protein, skin changes) syndrome
• Hyperviscosity: usually when IgM >4 g/dL, IgG >5 g/dL, or IgA >7 g/dL
• Coagulopathy: seen in amyloid due to binding & depletion of Factor X
• AL amyloidosis (see “Amyloidosis”)
Diagnostic and staging evaluation (Lancet Onc 2014;15:e538)
• Dx: BM plasma cells ≥10% or bx-proven plasmacytoma & ≥1 MM-defining event:
(a) myeloma-related organ or tissue impairment (ROTI) = lytic bone lesions, Ca >11 mg/dL, Cr >2 mg/dL, or Hb <10 g/dL
(b) any of the following biomarkers: BM plasma cells ≥60%, serum free light chain (FLC) ratio ≥100:1, >1 focal lesion on MRI
• Variants
Solitary bone or extramedullary plasmacytoma: no periph plasmacytosis or other ROTI
Plasma cell leukemia: plasma cell count >2000/µL in peripheral blood
Nonsecretory MM (~2% of MM Pts; MM without M protein)
• ✓ Peripheral smear for rouleaux (see insert), Ca, alb, Cr; ↓ anion gap, ↑ globulin, ↑ ESR
• Protein electrophoresis and immunofixation
Serum protein electrophoresis (SPEP): quantitates M component; ⊕ in >80% of Pts
Ddx of M component: MM, MGUS (vide supra), CLL, lymphoma, sarcoidosis, RA Polyclonal hypergam seen in inflammatory states: HIV, rheumatic diseases, cirrhosis
Urine protein electrophoresis (UPEP): detects Pts who secrete only light chains (= Bence Jones proteins), which are filtered rapidly from the blood
Immunofixation: shows component is monoclonal and identifies Ig type → IgG (50%), IgA (20%), IgD (2%), IgM (0.5%), light chain only (20%), nonsecretors (<5%)
Serum FLC assay: important for dx (esp. light chain-only Pts) and f/up response to Rx
• β2-microglobulin (β2M) and LDH levels reflect tumor burden
• BM bx cytogenetics: normal karyotype better than abnl. Standard risk = hyperdiploidy or t(11;14); high risk = hypodiploidy, del. 17p13 (~10% of Pts), t(4;14) & t(4;16).
• Skeletal survey (plain X-rays) to identify lytic bone lesions and areas at risk for pathologic fracture. Whole-body PET-CT (scalp to toe) or MRI often used to detect bone lesions.
Multiple Myeloma Staging Systems (OS does not account for cytogenetics) |
|||
Stage |
ISS Criteria* |
Durie-Salmon (DS) Criteria |
ISS Median OS |
I |
β2M <3.5 mg/L and albumin >3.5 g/dL |
All: Hb >10 g/dL; Ca ≤12 mg/dL; 0–1 lytic bone lesions; IgG <5 g/dL or IgA <3 g/dL or urine light chain <4 g/24 h |
62 mo |
II |
Fulfilling criteria for neither I nor III |
44 mo |
|
III |
β2M >5.5 mg/L |
Any: Hb <8.5 g/dL; Ca >12 mg/dL; >5 lytic bone lesions; IgG >7 g/dL or IgA >5 g/dL or urine light chain >12 g/24 h |
29 mo (30 mo if Cr <2 mg/dL; 15 mo if Cr ≥2 mg/dL) |
*Consider R-ISS incl chrom abnl & LDH (JCO 2005;23:3412 & 2015;61:2267).
Treatment and prognosis (NEJM 2016;375:754 & 1319; 2018;378:518 & 379:1811)
• Decisions dictated by risk stratification and transplant eligibility; generally incurable. Stratify via ISS criteria; R-ISS which includes chrom abnl & LDH (JCO 2015;61:2267).
• Rx incl. proteasome inhibitors: bortezomib (V), carfilzomib (K), ixazomib (I); immunomodulators: lenalidomide (R), thalidomide (T), pomalidomide (P); immunotherapy: daratumumab (anti-CD38, Dara), elotuzumab (anti-SLAMF7, Elo)
Other active drugs: dexamethasone (D), melphalan, panobinostat, cyclophosphamide
• Induction Rx w/ best response rate: proteasome inhib (V or K) + immunomod (eg, R). Triplet Rx ↑ OS vs. double (Lancet 2017;389:519). RVD most common regimen in US; KRD if high-risk (NEJM 2014;371:906 & 2016;374:1621). Dara-RD an option (NEJM 2019;380:2104).
Quad Rx (Dara-RVD) may improve response rates (Blood 2020;136:936).
• If not transplant eligible: induction Rx ↑ survival, not curative; consider maint chemo
• If transplant eligible: after induction chemo then high-dose melphalan + auto-HSCT. Not curative, but ↑ progression-free survival (PFS) vs. chemo alone (NEJM 2014;371:895, Lancet Onc 2015;16:1617). Offer if good perf. status & no prohibitive comorbid. Maint Rx w/ R improves PFS/OS (NEJM 2014;371:10). Timing of HSCT (upfront vs. relapse) debatable.
• Relapsed/refractory: HSCT (if good prior response, no prior HSCT), Elo-PD, Dara-PD, venetoclax (anti-Bcl-2) + dex in t(11;14); rarely use Allo-SCT. CAR-T w/ idecabtagene vicleucel (anti-B-cell maturation antigen) after >3 prior Rx: response rates >70% (NEJM 2019;380:1726 & 2021;384:705).
• Local radiation for solitary or extramedullary plasmacytoma
• Adjunctive Rx: bone: bisphosphonates (JCO 2007;25:2464), XRT for sx bony lesions
renal: avoid NSAIDs & IV contrast; consider plasmapheresis for acute renal failure
hyperviscosity syndrome: plasmapheresis; infxns: consider IVIg for recurrent infections
• Common toxicities of Rx: melphalan → myelosuppression; lenalidomide → low plts & thromboembolism; bortezomib → periph. neuropathy; steroids → hyperglycemia, infxn
WALDENSTRÖM’S MACROGLOBULINEMIA (WM)
Definition (Blood 2009;114:2375; NEJM 2012;367:826)
• B-cell neoplasm (lymphoplasmacytic lymphoma) that secretes monoclonal IgM
• 91% w/ MYD88 (NF-κB pathway) L265P mut., may distinguish from MM
• No evidence of bone lesions (IgM M component + lytic bone lesions = “IgM myeloma”)
Clinical manifestations
• Fatigue from anemia is most common sx
• Tumor infiltration: BM (cytopenias), hepatomegaly, splenomegaly, lymphadenopathy
• Circulating monoclonal IgM
Hyperviscosity syndrome (~15%): Neurologic: blurred vision (“sausage” retinal veins), HA, dizziness, Δ MS. Cardiopulmonary: congestive heart failure, pulm. infiltrates.
Type I cryoglobulinemia → Raynaud’s phenomenon
Platelet dysfxn → mucosal bleeding
• IgM deposition (skin, intestine, kidney); amyloidosis and glomerulopathy
• Autoantibody activity of IgM: Chronic AIHA (prominent rouleaux; 10% Coombs’ ⊕ = AIHA). Peripheral neuropathy: may be due to IgM against myelin-associated glycoprotein.
Diagnostic evaluation
• SPEP + immunofixation with IgM >3 g/dL; 24-h urine for UPEP (only 20% have ⊕ UPEP)
• Bone marrow biopsy: ↑ plasmacytoid lymphocytes; β2-microglobulin for prognostic eval
• Relative serum viscosity: ratio serum viscosity to H2O (nl 1.8); hyperviscosity when >5–6
Treatment
• Hyperviscosity: plasmapheresis; limit transfusions if possible (worsen hyperviscosity)
• Sx (eg, prog. anemia): ritux ± chemo (eg, bendamustine, Cy, etc.); ritux + ibrutinib (NEJM 2018;378:2399); zanubrutinib (Lancet Haem 2020;7:e837); everolimus or HSCT in salvage
HEMATOPOIETIC STEM CELL TRANSPLANTATION (HSCT)
Transplantation of donor pluripotent cells that can reconstitute all recipient blood lineages
Categories of Stem Cell Transplantation |
||
Feature |
Allogeneic (Allo) |
Autologous (Auto) |
Donor–recipient relationship |
Immunologically distinct |
Donor is also recipient |
Graft-vs.-host disease |
Yes |
No |
Graft-vs.-tumor effect |
Yes |
No |
Risk of graft contam. w/ tumor |
No |
Yes |
Relapse risk (leukemia) |
Lower |
Higher |
Transplant-related mortality |
Higher |
Lower |
• Types of Allo HSCT: based on donor/recipient matching of major HLA antigens on Chr. 6 (4 principal genes for serotyping: HLA-A, -B, -C, & -DR; each w/ 2 alleles ∴ 8 major Ag)
Matched related (MRD, sibling 8/8 major Ag match): lowest GVHD; preferred donor
Matched unrelated (MUD): ↑ risk of GVHD; ∴ matching of 10 HLA alleles (DQ also) to ↓ risk; chance of match correlates w/ ethnicity (NEJM 2014;371:339)
Mismatched related (eg, 1/8 Ag mismatch): ↑ available donor pool, but ↑ GVHD, rejection; ∴ need additional immunosuppression
Haploidentical: typically, between parents and young children or sibs (“half” match); early post-tx cyclophosphamide reduces GVH by destroying proliferating alloreactive T-cells
Umbilical cord blood: HSC processed at birth & stored. Yields lower cell number. Need 2 cords per adult. Neonatal immune cells: HLA-mismatch tolerated better, ↓ GVHD, slow immune reconstitution → ↑ late viral infections (Blood 2010;116:4693).
• Graft-vs.-host disease (GVHD): undesirable side effect of allo HSCT allogeneic T cells view host cells as foreign; ↑ incid. w/ mismatch or unrelated donors
• Graft-vs.-tumor (GVT): desired effect in allo-SCT; graft T cells attack host tumor cells
Indications (BBMT 2015;21:1863; BMT 2015;50:1037)
• Malignant disease:
Auto HSCT allows high-dose myeloablative chemo and then rescue what would be otherwise lethal cytopenias with autologous stem cells. Used in chemosensitive diseases such as relapsed/refractory DLBCL, MM, testicular germ cell tumor.
Allo HSCT produces graft-vs.-tumor (GVT) effect, in addition to hematopoietic rescue (used for AML, ALL, MDS, CML-blast crisis, CLL, lymphoma)
• Nonmalignant disease: allo HSCT replaces abnl lymphohematopoietic system w/ one from nl donor (eg, immunodeficiencies, aplastic anemia, hemoglobinopathies)
Transplantation procedure (for Allo HSCT)
• Pre-Tx conditioning regimen goal: immunosuppression to allow donor cell engraftment & anti-tumor efficacy to ↓ relapse risk. Type and dose of agents determine this balance.
Myeloablative conditioning: high-dose chemo and/or total body irradiation. Low relapse rates, high immunosuppression, higher transplant-related morbidity.
Reduced-intensity conditioning (“RIC”): lower dose of chemo → ↓ transplant-related morbidity/mortality, but ↑ relapse b/c it relies more on GVT effect (Blood 2015;126:23). Allows allo HSCT for older adults (>60) or Pts w/ comorbidities.
• Sources of hematopoietic stem cells (NEJM 2012;367:1487)
Bone marrow (BM): original source, preferred in non-malignant disease, ↓ GVHD rates
Peripheral blood stem cells (PBSC): easier to collect, more commonly used. BM vs. PBSC ≈ survival; BM ↓ chronic GVHD, PBSC ↓ graft failure, faster engraftment.
Umbilical cord blood stem cells (UCB): see above in Types of Allo HSCT
• Engraftment: absolute neutrophil count (ANC) recovers to 500/µL w/in ~2 wk w/ PBSC, ~2.5 wk w/ BM, ~4 wk w/ UCB. G-CSF accelerates recovery by 3–5 d in all scenarios.
Complications
• Either direct chemoradiotoxicities associated with preparative regimen or consequences of interaction between donor and recipient immune systems
• Engraftment syndrome: fever, rash, noncardiogenic pulm. edema, abnl LFTs, AKI, wt gain. Dx of exclusion: r/o infection, GVHD; Rx w/ 1 mg/kg steroids, rapid taper over 3–4 d.
• Sinusoidal obstruction syndrome (SOS): incidence ~10%, mortality ~30%
Previously known as veno-occlusive disease (VOD) (BBMT 2016;22:400). Mechanism: direct cytotoxic injury to hepatic venules → in situ thrombosis.
Symptoms: tender hepatomegaly, ascites, jaundice, fluid retention with severe disease → liver failure, encephalopathy, hepatorenal syndrome
Diagnosis: ↑ ALT/AST, ↑ bilirubin; ↑ PT with severe disease; Doppler U/S may show reversal of portal vein flow; ↑ hepatic wedge pressure; abnl liver bx
Treatment: supportive; prophylaxis with ursodiol; treat w/ defibrotide (Blood 2016;127:1656)
• Idiopathic pneumonia syndrome (IPS): 5–25% of Pts, >50% mortality (Blood 2003;102:2777)
Alveolar injury 2/2 direct toxicity → fever, hypoxia, diffuse infiltrates; occult infxn frequent
• Bleeding: incidence ~25%, 2/2 ↓↓ plts; sites: CNS, GI, pulm.; Ppx w plt transfusions
• Diffuse alveolar hemorrhage (DAH): Dx: bronchoscopy to exclude infection; ↑ bloody lavage fluid. Rx: 500–1000 mg Solu-Medrol × 3 d ± etanercept (BBMT 2015;1:67).
• Acute GVHD (usually within 6 mo of transplant; NEJM 2017;377:2167)
Clinical grades I–IV based on scores for skin (severity of maculopapular rash), liver (bilirubin level) and GI (volume of diarrhea); bx supports diagnosis
Prevention: immunosuppression (MTX + cyclosporine, or tacrolimus), T-cell depletion of graft, post-Tx cyclophos. for haploidentical, some high-risk allo (BBMT 2008;14:641)
Treatment: grade I → topical Rx; grades II–IV → associated with ↓ survival and ∴ treated with immunosuppressants (corticosteroids, CsA, tacrolimus, rapamycin, MMF)
• Chronic GVHD (developing or persisting >3 mo posttransplant; NEJM 2017;377:2565)
Clinical: malar rash, sicca syndrome, arthritis, obliterative bronchiolitis, bile duct degeneration, cholestasis, and many others. More common w/ PBSC than BM.
Treatment: immunosuppression; rituximab; photopheresis; ibrutinib (Blood 2017;130:21), ruxolitinib (NEJM 2021; 385:228), belumosudil (Blood 2021;138:2278)
• Graft failure
Primary = persistent neutropenia without evidence of engraftment
Secondary = delayed pancytopenia after initial engraftment; either immune mediated via immunocompetent host cells (graft rejection) or non–immune mediated (eg, CMV)
• Infectious complications
due to regimen-induced pancytopenia and immunosuppression
auto HSCT recipients: no immunosuppression ∴ at ↑ risk only pre-/postengraftment
both primary infections and reactivation events occur (eg, CMV, HSV, VZV)
Timing of Complications Following Allogeneic HSCT |
|||
|
Time After Transplant and Associated Risk Factors |
||
|
Days 0–30 Mucositis; organ dysfunction; neutropenia |
Days 30–90 Acute GVHD; ↓ cell immunity |
>90 Days Chronic GVHD; ↓ cellular & humoral immunity |
Viral infection |
Respiratory and enteral viruses, BK virus |
||
HSV* |
CMV*, HHV 6 & 7 |
||
|
EBV-related lymphoma |
||
|
|
VZV*, JC |
|
Bacterial infection |
Gram ⊕ cocci (Staph., Strep. viridans) GNRs (Enterobacteriaceae, Pseudomonas, Legionella, S. maltophilia) |
Encapsulated bacteria |
|
Fungal infection |
Candida spp. |
|
|
Aspergillus spp. |
|||
Parasitic infection |
|
T. gondii P. carinii S. stercoralis |
T. gondii P. carinii |
Regimen-related |
Pancytopenia |
|
Growth failure |
Mucositis, rash, alopecia |
Hypogonadism/infertility |
||
Nausea, vomiting, diarrhea |
Hypothyroidism |
||
Peripheral neuropathies |
Cataracts |
||
Hemorrhagic cystitis |
Avascular necrosis of bone |
||
Veno-occlusive disease |
2nd malignancy |
||
IPS/Interstitial pneumonitis |
|
||
Immune-mediated |
Acute GVHD |
Chronic GVHD |
|
Primary graft failure |
Secondary graft failure |
||
*Primarily among persons who are seropositive before transplant.
Prophylaxis/Supportive Medications During HSCT |
||
Medication |
Prophylaxis Against |
Duration |
Fluconazole or posaconazole |
Candida |
75 d |
Acyclovir |
HSV/VZV |
365 d |
Valganciclovir, ganciclovir, or letermovir if CMV ⊕ |
CMV |
100 d or when no longer immunosuppressed |
Antibiotics (eg, fluoroquinolone) |
Bacterial infxn |
While neutropenic |
TMP-SMX or atovaquone |
PCP |
365 d or when off immunosupp. |
Allopurinol |
Hyperuricemia |
Until d –1 |
LUNG CANCER
Epidemiology and risk factors
• Most common cause of cancer-related death for both men and women in the U.S.
• Two main types: non-small cell (NSCLC, ~85% of cases); small cell (SCLC, ~15%)
• NSCLC comprised of adeno (40%), squamous cell (SCC, 20%), & large cell (5%) carcinomas, as well as other / not classified (20%)
• Cigarette smoking: 85% of lung cancers occur in smokers; risk ∝ total pack-yrs, ↓ risk after quitting/reducing but not to baseline (Int J Cancer 2012;131:1210).
SCC & SCLC almost exclusively in smokers, adeno most common type in nonsmokers.
• Asbestos: when combined with smoking, synergistic ↑ in risk of lung cancer
• Other: RT (for other cancer); HIV; environ. toxins (radon, 2nd-hand smoke); pulm. fibrosis
Screening (JAMA 2021;325:962)
• Annual low-dose chest CT in ≥20 pack-year current or former (quit w/in 15 y) smokers, age 50–80 y → 20% ↓ lung cancer-related mortality (NEJM 2020;382:503)
• High rates of screen-detected nodules. Multidisciplinary mgmt recommended (pulm., med onc, thoracic radiology & surgery; NCCN Guidelines: Lung Cancer Screening v.1.2022).
Clinical manifestations
• ~10% asymptomatic at dx, detected incidentally (only 16% w/ localized dis. at presentation)
• Endobronchial growth of 1° tumor: cough, hemoptysis, dyspnea, pain, wheezing, post-obstructive pneumonia; more common with squamous or small cell (central location)
• Regional spread: can cause dysphagia (esophageal compression), stridor (tracheal obstruction), hoarseness (recurrent laryngeal nerve palsy). Pleural effusions & pleural metastases specifically = metastatic disease = Stage IV.
Pancoast’s syndrome: apical tumor → brachial plexus involvement (C8, T1, T2) → Horner’s syndrome, shoulder pain, rib destruction, atrophy of hand muscles
SVC syndrome (NEJM 2007;356:1862): central tumor → SVC compression → face/arm swelling (>80%), neck/chest vein distention (~60%), SOB/cough (~50%), HA (~10%); Rx = steroids, diuretics, RT ± chemo, SVC stent if severe sx, anticoag if clot
• Extrathoracic metastases: brain (~25% of patients at dx), bone, liver, adrenal
• Paraneoplastic syndromes
Endo: SIADH (SCLC); ACTH (SCLC) → Cushing’s; PTH-rP (SCC) → hyperCa
Skeletal: digital clubbing (NSCLC), hypertrophic pulm. osteoarthropathy (adenocarcinoma) = symmetric polyarthritis and proliferative periostitis of long bones
Neuro (SCLC): Eaton-Lambert (anti-P/Q-type voltage-gated Ca2+ channel Abs), peripheral neuropathy (anti-Hu, anti-PCA-2, anti-CRMP5), cerebellar degeneration (anti-Hu, anti-Yo, anti-Ri, anti-Tr), encephalomyelitis (anti-Hu, anti-Ma1/2, anti-CRMP5)
Hematologic: hypercoagulable state (adenocarcinoma), DIC
Diagnostic and staging evaluation (NCCN Guidelines v.7.2021)
• Staging: based on tumor size and extent of invasion (T), regional LN involvement [N: N0 (none), N1 (ipsilat. hilar), N2 (ipsilat. mediast.), N3 (contralat., supraclav.)] and presence of metastases (M) (Chest 2017;151:193). Pleural effusions/implants = Stage IV.
5-y survival: ~70–90% for stage I, 50–60% stage II, 15–35% stage III, 0–10% stage IV (J Thorac Oncol 2016;11:39); survival improving with newer therapies (NEJM 2020;383:640).
• Imaging: CT w/ contrast of chest & abd/pelvis to assess for mets (adrenal, liver, bones).
PET-CT more Se than CT alone for detecting mediastinal and distant mets as well as
bone mets (NEJM 2009;361:32). Brain MRI for all Pts except stage IA.
• Pathology: via bronchoscopy (central lesions) or CT-guided needle bx (peripheral lesions or accessible sites of suspected metastasis); mediastinoscopy (LN bx / eval), VATS (eval. of pleural peripheral lesions), thoracentesis if pleural effusion (cytology).
• Genetics (usually just in adv/metastatic disease): ✓ EGFR (include in stage IIA-IIIB), ALK, ROS1, MET, KRAS, RET, BRAF, NTRK fusion for all non-squamous disease (incidence lower in SCC but still consider testing); PD-L1 expression
• PFTs w/ quant V/Q if Rx includes resection; need 30% nl predicted lung fxn after resection
NSCLC Treatment (NCCN Guidelines v.7.2021; Lancet 2021;398:535)
• Localized disease: surgery if possible; RT (eg, stereotactic ablative radiotherapy [SABR]) vs. chemo/RT if not (see Figure 5-7)
• Metastatic disease: >20 new Rx since 2015. Some targeted Rx substantially improves survival & are better tolerated (see Stage IV Rx table); however, 5-y survival still poor.
First-line immunotherapy (IO) ± chemo for most Pts w/o targetable mutations.
Pemetrexed + pembrolizumab often continued as maintenance when used.
Palliative care ↑ survival (NEJM 2010;363:733); palliative RT for local sx from tumor/mets.
• Avoid combining IO & targeted Rx, ↑↑ tox (eg, rash/diarrhea, lung/liver injury [can be fatal])
• Solitary brain metastasis (oligomet): surgical resection + brain radiation may ↑ survival
(NCCN Guidelines v7.2021; NEJM 2017:377:1919, 2018;378:113, & 2020;383:1711; Lancet 2021;398:1344)
Stage IV NSCLC Treatment |
|||
Type |
Genetics |
% |
Treatment |
Adeno or Other |
EGFR |
~15 |
1st line: osimertinib if L858R, ex19del (NEJM 2018;378:113) |
EGFR ex20 insertion |
2–3 |
Subsequent Rx w/ amivantamab (JCO 2021;39:3391) or mobocertinib (JAMA Onc 2021;7:e214761) after platinum chemo |
|
ALK |
~4 |
1st line: alectinib pref. b/c CNS activity (NEJM 2017;377:829) |
|
ROS1 |
2 |
Crizotinib pref. (JCO 2017;35:2613); entrectinib (Lancet Onc 2020;21:261) |
|
MET ex14 skip or amplif. |
3 |
Capmatinib (NEJM 2020;383:944) or tepotinib (NEJM 2020;383:931) |
|
RET |
1 |
Selpercatinib (NEJM 2020;383:813) or pralsetinib (Lancet Onc 2021;22: 959-969) |
|
BRAF V600E |
1–3 |
Dabrafenib + trametinib (Lancet Onc 2016;17:984) |
|
NTRK fusions |
<1 |
Larotrectinib (NEJM 2018;378:731) |
|
KRAS G12C |
~13 |
Subsequent Rx w/ sotorasib (NEJM 2021;384:2371) |
|
PD-L1≥50% |
30 |
Pembrolizumab or chemo + pembro (NEJM 2018;378:2078) |
|
No targets & PD-L1 <50% |
|
Chemo (carbo/pemetrexed) + pembro (NEJM 2018;378:2078) or carbo/paclitaxel + anti-VEGF Ab (bevacizumab) + atezo (NEJM 2018;378:2288) |
|
Squam |
PD-L1 ≥50% |
|
Pembrolizumab or chemo/pembro (NEJM 2016;375:1823) |
|
PD-L1 <50% |
|
Chemo [carbo/paclitaxel] + pembro (NEJM 2018;379:2040) |
Many are specific tyrosine kinase inhibitors (‘tinibs) directed against EGFR, ALK, etc. Amivantamab is an EGFR- MET bispecific mAb. Pembrolizumab is a mAb against PD-1 on lymphocytes (ie, an immune checkpoint inhibitor).
SCLC staging and treatment (NCCN Guidelines v.1.2022)
• SCLC usually disseminated at presentation; poor overall survival
• Can be very responsive to chemotherapy, but rapidly develop resistance
• Diagnosis: all have Rb & p53 mutations (Nature 2015;524:47); 90% w/ neuroendo markers
• Treatment: primarily chemo (platinum + etoposide); adding anti-PD-L1 Ab (atezolizumab)
↑ survival (NEJM 2018;379:2220), as does concurrent thoracic RT in limited-stage disease

• Prophylactic cranial irradiation (PCI) controversial. Offered to potentially ↑ survival for LS-SCLC in complete remission (NEJM 1999;341:476). Role in ES-SCLC being studied.
• Second line: low response rates, short survival. Options: lurbinectedin, single-agent chemo (eg, topotecan, docetaxel), reRx w/ platinum doublet, anti-PD-1 (eg, nivo).
BREAST CANCER
Epidemiology
• In U.S., most common cancer in women; 2nd leading cause of cancer death in women
• Genetic risk: 15–20% ⊕ FHx → 2× ↑ risk; ~45% familial cases a/w germline mutation.
BRCA1/2: 35–85% lifetime risk of breast ca. Germline loss-of-function mutations in PALB2 a/w 35% ↑ risk breast cancer by age 70. Moderate risk mutations CHEK2 and BARD1 a/w 15–30% lifetime risk of breast cancer (NEJM 2021;384:428).
• Estrogen: ↑ risk with early menarche, late menopause, late parity or nulliparity (NEJM 2006;354:270); ↑ risk with prolonged HRT (RR = 1.24 after 5.6 y; JAMA 2003;289:3243); OCP use a/w extremely low to no ↑ risk (NEJM 2017:317:2228; JAMA Oncol 2018;4:516)
• Benign breast conditions: ↑ risk if atypia (atypical ductal or lobular hyperplasia; NEJM 2015;372:78) or proliferative (ductal hyperplasia, papilloma, radial scar, or sclerosing adenosis) features; no ↑ risk w/ cysts, simple fibroadenoma, or columnar changes
• ↑ risk with h/o ionizing radiation to chest for treatment of Hodgkin lymphoma
Prevention (if high-risk: eg, FHx, LCIS, atypical hyperplasia)
• Tamoxifen (contraindic. in preg): ↓ risk contralateral breast ca as adjuvant Rx. Approved for 1° prevent. if ↑ risk: ↓ invasive breast cancer, but ↑ DVT & uterine ca.
• Raloxifene (only if post-menopausal): ↓ risk of invas breast ca, vertebral fx, & uterine ca, ↑ risk stroke & DVT/PE (NEJM 2006;355:125); less effective than tamoxifen for prevention.
• Aromatase inhib. (post-menopausal): ↓ risk >50% (Lancet 2014;383:1041), ↑ osteoporosis
• BRCA1/2 ⊕: intensified surveillance vs. prophylactic bilat. mastectomy which ↓ risk ~90%; bilat. salpingo-oophorectomy ↓ risk of ovarian and breast cancer (NEJM 2016;374:454)
Clinical manifestations
• Breast mass (hard, irregular, fixed, nontender), nipple discharge (higher risk if unilateral, limited to 1 duct, bloody, associated with mass)
• Special types: Paget disease → unilateral nipple eczema + nipple discharge; inflammatory breast cancer → skin erythema and edema (peau d’orange)
• Metastases: lymph nodes, bone, liver, lung, brain
Screening (JAMA 2015;314:1599; Annals 2019;170:547)
• Mammography: ~20–30% ↓ in breast cancer mortality, smaller abs. benefit in women <50 y (JAMA 2018;319:1814); digital breast tomosynthesis (3-D) ↑ specificity (JAMA Oncol 2019;5:635); suspicious findings: clustered microcalcifications, spiculated, enlarging
• ACS: annual mammo starting at 45 (consider biennial after 54), cont. if life expect ≥10 y
• USPSTF: screen biennially ages 50–75 (some may want to begin at 40)
• ↑ risk: screen earlier w/ exam and mammo (age 25 in BRCA1/2 carrier, 5–10 y before earliest FHx case, 8–10 y after thoracic RT, upon dx of ↑ risk benign disease)
• MRI: superior to mammo in high-risk and young Pts; consider annually if >20% lifetime risk (eg, ⊕ FHx, BRCA1/2, prior chest RT) (Lancet 2011;378:1804)
• Germline genetic testing: if metastatic, TNBC, ♂, age ≤45y, or by FHx (NCCN v1.2022)
Diagnostic evaluation
• Palpable breast mass: age <30 y → observe for resolution over 1–2 menstrual cycles; age <30 y, unchanging mass → U/S → aspiration if mass not simple cyst;
age >30 y or solid mass on U/S or bloody aspirate or recurrence after aspiration → mammo (detect other lesions) and either fine-needle asp. or core-needle bx
clearly cancerous on exam or indeterminate read or atypia on bx → excisional bx
• Suspicious mammogram with normal exam: stereotactically guided bx
• MRI: detects contralateral cancer in 3% of Pts w/ recently dx breast cancer & contra-lateral mammo (but PPV only 21%) (NEJM 2007;356:1295); utility remains unclear
Staging
• Anatomic: tumor size, chest wall invasion, axillary LN mets (strongest prognostic factor)
• Histopathologic: type (little prognostic relevance) & grade; lymphatic/vascular invasion
In situ carcinoma: no invasion of surrounding stroma
Ductal (DCIS): ↑ risk of invasive cancer in ipsilateral breast (~30%/10 y)
Lobular (LCIS): benign entity; marker of ↑ risk of inv. cancer in either breast (~1%/y)
Invasive carcinoma: infiltrating ductal (70–80%); invasive lobular (5–10%); tubular, medullary and mucinous (10%, better prognosis); papillary (1–2%); other (1–2%)
Inflammatory breast cancer (see above): not a histologic type but a clinical reflection of tumor invasion of dermal lymphatics; very poor prognosis
Paget disease (see above): ductal cancer invading nipple epidermis ± associated mass
• Tissue biomarkers: estrogen and progesterone receptor (ER/PR), HER2
• Oncotype DX 21-gene expression recurrence score predicts which ER ⊕, HER2 ⊖ will have minimal benefit from adjuvant chemo in LN ⊖ (NEJM 2018;379:111) and LN ⊕ (1–3) disease (NEJM 2021;385:2336)
General Approach to Treatment (JAMA 2019;321:288 & 1716) |
|
DCIS |
Mastectomy or lumpectomy ± RT ± chemoprevention (Lancet 2016;387:849 & 866) |
I |
Surgery + RT |
II |
+ adjuv. chemo if ↑ risk: tumor >2 cm or ⊕ LN or triple ⊖ or Oncotype DX-guided + hormonal Rx for ER/PR ⊕: add ovarian suppression if ↑ risk (NEJM 2018; 379:122) + anti-HER2 Rx and chemo if HER2 ⊕ and tumor ≥1 cm or ⊕ LN |
III |
Neoadjuvant chemo → surgery + RT ± adjuvant chemotherapy + hormonal Rx for ER/PR ⊕: add ovarian suppression if premenopausal + anti-HER2 Rx for HER2 ⊕: usually trastuzumab + pertuzumab |
IV |
ER/PR ⊕: combined aromatase & CDK4/6 inhibitors (NEJM 2016; 375:1925) ER/PR ⊖/HER2 ⊕: chemo + anti-HER2 therapy Triple ⊖: chemo ± immune checkpoint inhibitor Bony mets: bisphosphonates & denosumab ↓ fractures (Cochrane 2017;CD003474) |
Surgery and Radiation for Local Control |
|
Intervention |
Indication |
Breast conserving |
Stage I–II, lumpectomy + sentinel lymph node biopsy* + RT |
Modified radical mastectomy |
Large tumor relative to breast, multicentric dis., prior chest RT, diffuse microcalcifications, ⊕ margins after lumpectomy |
Post mastectomy Radiation |
≥4 ⊕ LN, tumor >5 cm, ⊕ surgical margins, chest wall or skin involvement (Lancet 2014;384:1848) |
*Axillary lymph node dissection indicated for palpable axillary LNs
Systemic Therapy |
||
Indic. |
Class |
Examples |
ER/PR ⊕ (Lancet 2017;389: 2403) |
Endo (NEJM 2019;380: 1226) |
Tamoxifen: adjuvant Rx for low-risk pre-meno; ↓ recurrence & ↓ mortality; 10 y superior to 5 y (Lancet 2011;378:771 & 2013;381:805) Aromatase inhib (AI; anastrozole, letrozole, exemestane): adjuvant Rx for post-meno; ↑ OS vs. tam. (Lancet 2015;386:1341); 7 y of Rx ↑ DFS vs. 5 y of Rx (NEJM 2021;385:395) Adding selective ER degrader (fulvestrant) to AI ↑ OS if mets |
Ovarian suppress. |
LHRH agonists (eg, leuprolide) or oophorectomy: adjuvant Rx for high-risk pre-meno combined with tam. or AI (NEJM 2018;379:122) |
|
Cell prolif. (NEJM 2012;366: 520) |
CDK 4/6 inhib (eg, palbociclib, abemaciclib, ribociclib): + AI (1st-line metastatic Rx) or fulvestrant ↑ PFS (& OS for ribociclib) in stage IV vs. AI alone (NEJM 2018;379:1926; JCO 2017;35:3638); + AI for adjuvant (Ann Onc 2021;32:1571) mTOR inhib (everolimus): + AI (exemestane) ↑ OS in stage IV |
|
PIK3CA ⊕ |
PI3K inhib |
Alpelisib + fulvestrant ↑ PFS in met HR⊕ (NEJM 2019;380:1929) |
HER2 ⊕ (Lancet 2017;389: 2415) |
HER2- targeted |
Trastuzumab (anti-HER2): 1st-line Rx combined w/ chemo Trastuzumab emtansine (mAb linked to chemo): ↓ risk of recurrence/death if residual disease post neoadj. Rx (NEJM 2019;380:617); preferred 2nd line Rx for met. disease Trastuzumab deruxtecan (mAb linked to chemo): emerging data as 2nd line Rx for met disease, ↓ risk disease progression vs. trastuzumab emtansine, ↑ lung toxicity (NEJM 2022;386:1143) Margetuximab (anti-HER2): combined w/ chemo preferred after 2+ lines of Rx (JAMA Oncol 2021;7:573) |
Stage I–III (above) |
Chemo |
Neoadjuvant: to conserve breast & evaluate Rx efficacy, equivalent OS as adjuvant (JCO 2008;26:778) Adjuvant: use anthracycline ± taxane. Nb, endocrine only for some ER/PR ⊕ based on oncotype Dx (see staging). |
Triple ⊖ |
Immune |
Pembrolizumab (anti-PD-1 mAb) + chemo 1st-line for early stage (NEJM 2020;382:810) & PD-L1 ⊕ met dis. (Lancet 2020;396:1817) |
Ab-drug conjugate |
Sacituzumab govitecan: anti-trop-2 Ab linked to chemo ↑ PFS & OS in heavily pre-Rx’d metastatic disease (NEJM 2019;380:741) |
|
BRCA ⊕ |
PARP inh |
Olaparib & talazoparib (NEJM 2017;377:523 & 2018;379:753) |
DFS, disease-free survival; OS, overall survival; PFS, progression-free survival
PROSTATE CANCER
Epidemiology and risk factors (NEJM 2003;349:366)
• Most common cancer in U.S. men; 2nd most common cause of cancer death in men
• Lifetime risk of prostate cancer dx ~16%; lifetime risk of dying of prostate cancer ~3%
• ↑ risk with ↑ age (rare if <45 y), in African Americans, ⊕ FHx, BRCA mutations
Clinical manifestations
• Most prostate cancers (78%) are asymptomatic and localized at diagnosis
• Metastatic dis. sx primarily from bone mets: bone pain, spinal cord compression, cytopenias
Screening (JAMA 2014;311:1143; Lancet 2014;384:2027)
• PSA: 4 ng/mL cut point neither Se nor Sp; can ↑ with BPH, prostatitis, acute retention, after bx or TURP, and ejaculation (no significant ↑ after DRE, cystoscopy); 15% of men >62 y w/ PSA <4 & nl DRE have bx-proven T1 cancer (NEJM 2004;350:2239)
• Digital rectal exam no longer recommended due to limitations, no mortality benefit
• ACS rec: ≥50 y (or ≥45 y AA or ⊕ FHx) discuss PSA screening, informed decision making
• USPSTF (JAMA 2018;319:1901) rec discuss pros/cons w/ Pt (no ↓ in prostate ca-related mort.)
Diagnostic evaluation, staging, and treatment (NCCN Guidelines v1.2022)
• Transrectal ultrasound (TRUS) guided biopsy (6–12 cores)
• Multiparametric MRI (± endorectal coil): improves detection (NEJM 2018;378:1767)
• Gleason score & grade group (histology): Gleason score determines Grade Group. 1 = best → 5 = worst. Group 1: 3+3=6 (most common histologic pattern is 1st #, next is 2nd #), Group 2: 3+4=7, Group 3: 4+3=7, Group 4: 4+4=8, Group 5: all higher.
*In asx Pts w/ life expect ≤5 y & very low-to-intermed risk dis., no w/up or Rx until sx. NCCN Guidelines v1.2022
†RP more short-term impotence (unless ADT) & incont than RT; over yrs → similar (NEJM 2016;375:1425)
Treatment of Metastatic Prostate Cancer (NCCN Guidelines v1.2022) |
|
Androgen deprivation therapy (ADT) |
Prostate ca requires androgen signaling for growth. ADT backbone of Rx. Med: 1. Luteinizing hormone-releasing hormone (LHRH) agonist (eg, leuprolide) ± 1st-gen anti-androgen (nilutamide, bicalutamide), or 2. LHRH antagonist (degarelix) Surgery: bilateral orchiectomy |
Hormone- sensitive prostate cancer (HSPC) |
Def: ADT sensitive (ie, PSA ↓ w/ Rx): all prostate ca initially sensitive Workup/testing: PEx & PSA q 3–6 mos; sx-guided imaging Rx: 1. ADT alone (only if minimal disease, eg, rising PSA); or 2. ADT + abiraterone/pred or enza/apalutamide (↑ OS vs. ADT alone); or 3. Docetaxel + ADT (↑ OS vs. ADT alone, espec. in high-volume disease) |
Castration-resistant prostate cancer (CRPC) Always continue ADT |
Def: All met. HSPC eventually becomes CRPC (ie, progression despite castration androgen levels on ADT), due to re-estab. of androgen signaling via other mech. ∴ more potent anti-androgens are needed. Rx: New-gen. anti-androgens: abiraterone (biosynth inhib.) + pred, or enza/daro/apalutamide (receptor blocker) ↑ PFS & OS Targeted: BRCA1/2, ATM (homologous recomb genes, ~20%): olaparib; MSI-H/Lynch syndrome (2–5%): pembro; Cancer vaccine: Sipuleucel-T Chemo: doce/cabazitaxel +pred/dex+Luetitium-177 (NEJM 2021;385:1091) Bone-active agents: 1. denosumab or zoledronic acid ↓ skeletal-related events (SREs); 2. radium-223 used in bone-only dis, ↓ SREs & ↑ OS |
(NEJM 2017;377:338 & 352; 2019;381:121; 2019;381:13; Lancet 2016;387:1163)
Prognosis and monitoring
• PSA level, Gleason score/grade group and age are predictors of metastatic disease
• In surgically treated Pts, 5-y relapse-free survival >90% (excellent) if disease confined to organ, ~75% if extension through capsule, and ~40% if seminal vesicle invasion
• PSA q6mo for 5 y if curative Rx; consider PET Axumin scan if ↑ PSA ± equivocal bone scan
COLORECTAL CANCER (CRC)
Epidemiology and risk factors (CA Cancer J Clin 2018;68:7)
• 4th most common cancer in U.S. men & women; 2nd leading cause of all cancer death
• 90% of cases occur after age 50. ~75% are sporadic.
• IBD a/w ~0.3%/y ↑ risk of CRC. ↑ risk w/ ↑ extent and duration of disease.
Heritable Genetic Syndromes |
|||
Disorder |
CRC risk |
Pathophysiology |
Assoc Cancers |
Hereditary nonpolyposis colorectal cancer (HNPCC or Lynch) |
~80% lifetime |
Most common hered. CRC (~3%). Mismatch repair mut (eg, MSH2, MLH1). Dx: ≥3 family HNPCC cancer, 1 dx before 50 y, involves 2 gen. Typically right-sided. |
Endometrial, ovarian, stomach, urothelial, small bowel & pancreas |
Familial adeno polyposis (FAP) |
100% lifetime |
Mutation in APC gene → 1000s of polyps at young age |
Thyroid, stomach, small bowel |
MUTYH-assoc. polyposis (MAP) |
40–100% lifetime |
Autosomal recessive; consider if mult. polyps but ⊖ for FAP |
Duodenal, ovarian, bladder, skin |
• ASA rec for 1° prev. if 50–59 y & ≥10% 10-y CRC risk. Must take for at least 10 years.
Screening (JAMA 2021;325:1965)
• Average-risk Pts: start at age 45, repeat q1–10y based on screening modality
• ↑ risk Pts: ⊕ FHx: screen age 40 or 10 y before index dx, then q5y. IBD: colo 8–10 y after dx, then q1–2y. Suspect familial syndrome: genetic counsel, screen age 20–25, yearly.
• Colonoscopy: preferred; 90% Se for lesions >1 cm. If polyp, re ✓ in 3–5 y. Adenomatous polyp removal a/w ↓ CRC mortality (NEJM 2012;366:687). Adenoma: ↑ risk of malig. if >2.5 cm, villous, or sessile; can progress to CRC over ~10 y interval (sporadic & familial).
• Sigmoidoscopy: benefit w/ 1-time flex-sig (Lancet 2017;389:1299); less Se than colo or CTC
• CT colonography (CTC): ~90% Se for lesions ≥1 cm but less if smaller (NEJM 2008;359:1207). If high-risk, Se only 85% for neoplasia ≥6 mm (JAMA 2009;301:2453).
• Combo DNA + Hb immunoassay w/ ~90% Se & Sp (NEJM 2014;370:1287)
• Occult blood (FOBT): use 3-card home testing (Se 24%) yearly
Clinical manifestations
• Distal colon: Δ bowel habits/ caliber, obstruction, colicky abd. pain, hematochezia
• Proximal colon: iron defic. anemia, dull vague abd pain, liquid stool
• Associated with Streptococcus bovis bacteremia and Clostridium septicum sepsis
Cancer genetics (Nature 2014;513:382)
• Microsatellite stable (MSS) vs. high instability (MSI-H): latter sign of mismatch repair gene failure, accounts for 15% CRC, presents more often as early stage, ~5% of met dis.
• Mutations: APC (~80%); KRAS (~40%); TP53 (50–70%); DCC, SMAD4, BRAF (~15%), HER2 amplification (<5%)
Staging and treatment (NCCN Clin Pract Guidelines; version 3.2021)
• TNM staging, including CT chest and abdomen/pelvis with contrast ± MRI pelvis for rectal
• Baseline CEA: monitor post resection or follow response; not for screening
• Chemo options (Lancet 2014;383:1490): 5-FU/leucovorin (LV) foundation. 5-FU/LV + oxaliplatin &/or irinotecan (FOLFOX, FOLFIRI, FOLFOXIRI, resp). Capecitabine oral 5-FU prodrug. TAS102 (trifluridine + tipiracil) in progressive disease (NEJM 2015;372:1909).
Anti-VEGF (bevacizumab) added to chemo ↑ OS in all subsets of mCRC.
• Targeted therapy: anti-EGFR mAb (cetuximab or panitumumab) in unmutated KRAS/NRAS/BRAF (NEJM 2013;369:1023), better for L-sided disease; encorafienib + cetuximab + binimetinib in BRAF V600E (NEJM 2019;381:1632)
• ImmunoRx: anti PD-1 (NEJM 2020;383:2207) ± CTLA-4 in MSI-H/ MMRd met CRC
• Radiation: loc. adv. rectal (T3/N+) & colon (unresectable); consider for oligo-mets
TNM |
Path. Criteria |
5-y Surv. |
Treatment |
I |
Submucosa/muscularis |
94–97% |
Surgery alone (resect & analyze ≥12 LN) |
IIA |
Serosa |
83% |
Surgery. Consider adjuvant chemo for high-risk Stage II: obstruction, perf, adherence, inadequate LN sampling (<12 LNs). |
IIB |
Peritoneum |
74% |
|
IIC |
Direct invasion |
56% |
|
IIIA |
≤6 ⊕ LNs |
86% |
Surgery + FOLFOX (6 mo) Pre RT ± chemo if rectal (NEJM 2006;355:1114) |
IIIB |
Varying # ⊕ LNs & local invasion |
51–77% |
|
IIIC |
15–47% |
||
IV |
Distant metastases* (NEJM 2014;371:1609) |
5% |
Chemo (FOLFOXIRI if high-risk) ± anti-PD-1 (MSI-H only) ± resect isolated mets |
*Oligometastatic dis (few, limited mets) sometimes still curable w/ chemo + metastatectomy/radiation
PANCREATIC TUMORS
Genetics and path (Nat Rev Dis Primers 2016;2:16022)
• Histologic types: adenocarcinoma (~85%), acinar cell carcinoma, endocrine tumors, cystic neoplasms (<10%); rarely, mets to pancreas (eg, lung, breast, renal cell)
• Location: ~60% in head, 15% in body, 5% in tail; in 20% diffuse infiltration of pancreas
• Adeno. mut.: KRAS (>90%), p16 (80–95%), p53 (50–75%), SMAD4 (~55%), BRCA (10%)
Epidemiology and risk factors (Lancet 2016;388:73)
• 4th leading cause of cancer death in U.S.; 80% panc adeno in ages 60–80 y; M>F (1.3:1)
• Acquired risk factors: smoking (RR ~1.5; 25% cases), obesity, chronic pancreatitis, T2DM
• Hereditary (5–10%): familial breast/ovarian CA (BRCA2); hereditary chronic pancreatitis (mutation in cationic trypsinogen gene (PRSS1, SPINK1); familial cancer syndromes: atypical multiple mole melanoma (CDKN2A/p16), Peutz-Jeghers (LKB1), ataxia-telang.
Clinical manifestations
• Painless jaundice (w/ pancreatic head mass), pain radiates to back, ↓ weight & appetite
• New-onset atypical DM (25%); migratory thrombophlebitis (Trousseau’s syndrome)
• Exam: jaundice, RUQ/epigastric nontender mass, palpable gallbladder (Courvoisier’s sign); hepatomegaly; ascites; L supraclavicular node (Virchow’s)
• Laboratory tests may show ↑ bilirubin, ↑ alk phos, anemia
Diagnostic and staging evaluation (NCCN Guidelines v.2.2021)
• Pancreatic protocol CT scan (I+ w/ arterial & venous phase imaging) or MRI w/ contrast
• If no lesion seen but concerning signs & sx as above → EUS, ERCP, or MRCP
• Biopsy pancreatic lesion via EUS-guided FNA (pref. for surgical candidates) or CT-guided (risk of seeding); if possible metastases, biopsy those for staging and dx.
• ✓ CA19-9 preop (nb, can be ↑ in benign liver/biliary dis.); may be useful to trend postop
• Germline genetic testing for all Pts with pancreatic cancer
Clinical (Radiologic) Staging Pancreatic Adenocarcinoma |
|
Resectable |
No extrapanc. dis. or bulky LAN; no arterial tumor contact [celiac axis (CA), SMA, common hepatic (CHA)]; and no venous contact [SMV, portal vein (PV)] or ≤180° + patent veins (ie, no tumor thrombus) |
Borderline resectable |
No extrapanc dis. or bulky LAN. Head/uncinate: contact w/ CHA (no extension to CA or HA bifurcation), SMA contact ≤180°, variant anatomy. Body/tail: contact CA ≤180° or >180° but w/o gastro-duodenal art. or aortic. Venous: SMV & PV contact ≤180° w/ contour irreg; contact w/ IVC. |
Unresect. |
Distant mets; or head/uncinate: contact >180° SMA, CA; or Body/tail: contact >180° SMA or CA; CA & aortic involvement; or Venous: SMV/PV involvement/not reconstructible |
Treatment of pancreatic adenocarcinoma (Lancet 2016;388:73)
• Resectable: pancreaticoduodenectomy (Whipple procedure) + adjuvant chemo:
modified FOLFIRINOX (5-FU + leucovorin, irinotecan, oxaliplatin) if ECOG 0-1
(NEJM 2018;379:2395), o/w gemcitabine + capecitabine (Lancet 2017;389:1011). Gemcitabine
monoRx recently standard, but now w/ ↓ role. Role of RT is controversial.
• Borderline: goal to ↓ tumor to allow complete resection (R0 – neg margin at histology) using neoadjuvant Rx (various approaches tested). General schema: chemo ± RT → restage & potential resection depending on response. May need vasc. reconstruction during resection. Regimens include: FOLFIRINOX; gemcitabine + nab-paclitaxel.
• Locally advanced (ie, unresectable): Rx is typically palliative. However, in highly select Pts recent trend toward Rx w/ FOLFIRINOX plus XRT followed by laparotomy for response assessment (imaging can be unreliable) and potential resection.
• Metastatic: clinical trials preferred; Rx based on performance status (PS)
Good PS: FOLFIRINOX, gemcitabine + nab-paclitaxel (NEJM 2013;369:1691); germline BRCA1/2 mut: maintenance olaparib (NEJM 2019;381:317) also ↑ response to platinum combination chemo (eg, cisplatin w/ gemcitabine), ICI for MSI-high.
Poor PS: gemcitabine; capecitabine; continuous infusion 5-FU
• Palliative care: Biliary/gastric outlet obstruct.: endoscopic stenting, IR drain, surg bypass. Pain: opiates, celiac plexus neurolysis, XRT. Wt loss: enzyme replacement.
Prognosis
• Resectable: if Rx’d w/ adjuvant FOLFIRINOX, 50+ mos, o/w ~30 mos
• Unresectable: if locally advanced ~1–2 y; if metastatic, ~1 y
Cystic lesions of the pancreas (Am J Gastroenterol 2018;113:464)
• Serous cystadenoma: usually benign; central scar or honeycomb appearance on imaging
• Mucinous cystic neoplasm (MCN): predominantly young females; multiloculated tumors in body or tail w/ ovarian-type stroma and mucin-rich fluid w/ ↑ CEA levels; precancerous
• Intraductal papillary mucinous neoplasm (IPMN): arises in main panc duct or branch
OTHER SOLID TUMORS
HEPATOCELLULAR CARCINOMA (HCC)
Risk factors (globally, 3rd leading cause of cancer death, espec. in Africa & Asia)
• Cirrhosis: present in 70–90% HCC cases
• Infectious: HCV & HBV (~75%), HBV/HDV coinfection; HBV can cause HCC w/o cirrhosis
• Toxic: EtOH (⅓ cases in U.S.), tobacco, aflatoxin from Aspergillus
• Metabolic disorders: NASH, DM, autoimmune hepatitis, hemochromatosis
Screening (screen high-risk Pts: cirrhosis, chronic HBV)
• Ultrasonography (U/S) + AFP q 6 mos; if high-risk may alternate U/S w/ MRI
• If lesion found or increasing AFP, perform 3-phase contrast CT or MRI
Clinical manifestations
• Exam: nonspec. c/w liver dysfxn (eg, hepatomegaly, ascites, jaundice, encephalopathy)
• Labs: coagulopathy, low albumin, elevated LFTs; r/o other etiologies of liver dysfxn
Diagnosis
• Can diagnose w/o biopsy in high-risk Pts with 3-phase contrast CT or MRI
• Only 15% of liver masses are HCC; liver metastases from other 1° more common
Treatment (NEJM 2019;380:1450)
• Localized disease (goal = cure) → resection if feasible (preferred), ablation, transarterial chemoembolization, or radiotherapy (SBRT) also options. Inadequate hepatic reserve → liver transplant if able (NEJM 1996;334:693); non-surgical local and/or systemic Rx if not.
• Systemic Rx: atezolizumab (anti-PD-L1) + bevacizumab preferred (NEJM 2020;382:1894), 2nd line: kinase (lenvatinib, sorafenib) or PD-(L)1 inhib (Lancet 2017;389:2492 & 2018;391:1163)
GASTRIC/ESOPHAGEAL CANCER
Epidemiology (Lancet 2017;390:2383 & 2020;29:635)
• Esophageal: Predominantly adenocarcinoma in U.S. (>60%), squamous cell (SCC) more common globally. Risk factors: GERD → Barrett’s esoph (adeno), smoking/EtOH (SCC)
• Gastric: Predominantly adenocarcinoma. Risk factors include H. pylori, FHx & cancer syndromes (eg, Lynch syndrome), nitrosamines in diet, smoking/EtOH.
Screening
• Esophageal: screen for Barrett’s w/ endoscopy if multiple risk factors (eg, hiatal hernia, age ≥50, male, chronic GERD, obese, smoking, FHx). Repeat q3–5y if Barrett’s.
• Gastric: typically not screened in U.S. More common in regions w/ higher incidence (eg, Japan, Korea, Chile) w/ endoscopy or barium radiographs.
Clinical manifestations
• Dysphagia, wt loss, early satiety, nausea, typically occult GI bleed, vague abd discomfort
• Exam: diffuse seborrheic keratoses (Leser-Trélat sign, though non-specific)
Diagnosis and staging
• Both diagnosed w/ endoscopic biopsy. CT chest/abd/pelvis for staging. If no mets → PET/CT (occult mets). If neg → endoscopic ultrasound for locoregional assessment.
• Staging laparoscopy needed for gastric cancer
• Molecular: ✓ HER2, PD-L1, microsatellite instability & mismatch repair
Treatment (NCCN Guidelines v5.2021 Gastric & v4.2021 Esophageal)
• If localized disease and good performance status/fit, goal is cure with surgery
• Localized: resection. Periop or neoadjuvant chemo/chemoRT if stage IB or higher (NEJM 2012;366:2074). Adjuvant nivolumab (NEJM 2021;384:1191).
• Advanced: sequential chemo w/ platinum/fluoropyrimidine (+ trastuzumab if HER2⊕, + nivolumab if PD-L1⊕), ramucirumab (VEGF, 2nd line), nivolumab/pembrolizumab (PD-1, 3rd line)
• Supportive care: pain/nausea mgmt. Consider G/G-J tube if obstruction, IVF prn.
MELANOMA
Clinical presentation and workup (NCCN Guidelines v1.2022)
• Suspicious skin lesion (ABCDE: asymm, border, color, diameter >6 mm, evolution) → bx
• Staging: I & II = no regional/distant mets. III = regional LN ⊕ or in-transit/satellite mets. Ulceration & thickness confer substage within I–III. IV = distant mets.
• Prognosis: excellent if localized dis., ≤1 mm thickness. Varies widely stage IIIA–IIID based on nodal burden (5yr OS 20–70%; ulceration, mitotic rate, & invasion depth predictive).
• Molecular diagnostics: check for BRAF V600E (present in ~50% of melanomas)
Treatment (NCCN Guidelines v1.2022; NEJM 2021;384:2229)
• Wide surgical excision + sentinel node mapping (unless in situ or low risk of node ⊕).
If sentinel node ⊕, nodal dissection vs. observation. No difference in survival w/ observation (Lancet Oncol 2016;17:757, NEJM 2017;376:2211).
• Checkpoint inhibitors (CPI = pembrolizumab, nivolumab, nivolumab/ipilimumab)
• BRAF/MEK inhibitors (dabrafenib/trametinib, vemurafenib/cobimetinib, encorafenib/binimetinib) if activating BRAF V600E mutation present
• Adjuvant for high-risk node ⊕ disease: CPI (↑ RFS, Lancet Onc 2020;21:1465 & JCO 2020;38:3925) or dabrafenib/trametinib (BRAF ⊕, ↑ OS, NEJM 2020;383:1139)
• Metastatic: CPI mono vs. dual (ipi+nivo a/w improved response rate especially for intracranial mets, NEJM 2018;379:722). Consider targeted Rx if BRAF ⊕.
OVARIAN CANCER
Histopathology
• Epithelial ovarian carcinoma (EOC, 95%): high-grade serous (most common, often dx at late stage, poor prognosis), low-grade serous, endometroid, clear cell, mucinous
• Non-epithelial (~5%): germ cell, sex cord-stromal. Rare: small cell, sarcoma.
Risk factors for EOC (Nat Rev Dis Primers 2016;2:16061)
• Nulliparity, early menarche, late menopause; EOC risk ↑ 2–7% each additional ovulatory yr
• Genetics: germline testing / genetics referral for all Pts (25% w/ heritable mutation), can inform Rx (eg, PARPi maintenance, JCO 2020;38:1222). 5% risk if one 1st-degree relative w/ ovarian cancer, 3.5% risk if one 2nd-degree relative (Obstet Gynecol 1992;80:700). ↑↑ risk w/ hereditary syndromes: BRCA1/2 (lifetime risk 17%/44%), Lynch syndrome (non-serous).
• History of pelvic radiation, endometriosis, or obesity (risk association less clear)
Clinical manifestations
• Nonspecific history of bloating, pelvic/abdominal pain, bowel sx, urinary sx
• Exam may reveal palpable pelvic mass, abdominal distention, or ascites
Diagnosis
• No screening modalities have shown survival benefit; no data to support at this time
• If concerning signs/sx (above), obtain pelvic U/S and/or CT A/P or pelvic MRI, CA-125
• Pathologic dx based on biopsy of lesion or sampling of ascitic fluid
Treatment
• Cytoreductive surgery: survival benefit even for stage IV and relapsed disease (Gynecol Oncol 2013;130:493; NEJM 2021;385:2123). Consider neoadj. chemo to ↑ likelihood of maximal cytoreduction at surgery and reduce surgical morbidity.
• Chemo: 1st line (neoadj., adj., adv.): carboplatin+paclitaxel (CP). PARPi maintenance (olaparib, niraparib, rucaparib; esp if homologous recombination deficient [eg, BRCA1/2]) or bevacizumab maintenance if homologous recombination proficient.
• Recurrence at >6 mos = platinum sensitive → Rx w/ platinum doublet. If <6 mos = platinum resistant → single agent chemo (eg, liposomal doxorubicin, gemcitabine) ± bevacizumab.
PRIMARY CNS TUMORS AND BRAIN METASTASES
Presentation and diagnosis
• Suspect if new focal neurologic deficits, seizure, memory or balance issues, or evidence of ↑ intracranial pressure (HA, n/v, vision change)
• MRI brain for suspected CNS tumors; CT less Se. Consider LP if c/f CNS lymphoma.
Primary CNS tumors
• Meningioma: histopath. grading (WHO grades). Grade 1 = benign, asx/small → observe. Large or sx → resection if able, radiation if not. Grade 2–3 → resection + adjuvant RT (defer if risk of RT complication high and tumor totally resected).
• Glioma: histopath. grading, Grade 1/2/3: astrocytoma, oligodendroglioma most common; Grade 4: glioblastoma. Grade 3/4 = “high grade” (Neuro Oncol 2021;23:1231).
• Notable testing: 1p/19q, IDH, MGMT methylation (predicts sensitivity to temozolomide)
• Treatment: max resection as feasible → adjuvant RT + temozolomide → surveillance. Steroids if neuro sx, hold until biopsy if c/f 1° CNS lymphoma. Anti-epileptics if seizures.
• Prognosis based on grade & histology. For glioblastoma, overall survival 12–15 mos, based on extent of tumor resection (J Neurosurg 2014;120:31).
Metastatic CNS Lesions
• ↑ incidence than 1° CNS. Most common: lung, breast, melanoma, RCC, gastro-esoph.
• Surgery if oligomet disease or symptomatic & resectable, steroids if neuro sx
• Radiation particularly if multiple mets or inoperable. Consider WBRT if many mets.
• Some systemic therapies (checkpoint inhibitors, targeted Rx in NSCLC) have CNS activity
IMMUNOTHERAPY & CELLULAR THERAPY
IMMUNE CHECKPOINT INHIBITORS (ICI)
Overview (Science 2018;359:1350)
• Immune checkpoints: co-inhibitory signaling molecules that limit immune responses
• Cytotoxic T-lymphocyte-assoc. protein 4 (CTLA-4): ↓ T cell activation via negative signals and ligand competition w/ CD28 (activating co-stim. molecule).
• Programmed cell death protein 1 (PD-1): on activated T cells, turns off T cell responses
• Prog. death-ligand 1 (PD-L1): PD-1 ligand expressed on tumor and/or immune cells
ICIs (Annals of Oncol 2017;28:2377) |
|||
Target |
Drugs |
Select Indications |
Common Adverse Events (approx. incidence, any grade) |
PD-1 |
Nivolumab Pembrolizumab Cemiplimab Dostarlimab |
Melanoma, NSCLC, RCC, HNSCC, esoph., endometrial, MSI-high tumors |
Rash (12%); hypothyroid (5%); pneumonitis (2%); colitis (<1%); hepatitis (<1%); type 1 DM (<1%) |
PD-L1 |
Atezolizumab Avelumab Durvalumab |
Urothelial carcinoma, NSCLC, SCLC, HCC (atezo) |
Rash (5%); hypothyroid (2%); adrenal insufficiency (<1%); colitis (<1%); pneumonitis (<1%) |
CTLA-4 |
Ipilimumab |
Melanoma, RCC (w/ PD1), mesothelioma (w/ PD1) |
Rash (21%); colitis (5%); hypophysitis (1%); hypothyroid (1%); pneumonitis (<1%); hepatitis (<1%) |
ICI toxicities (Nat Rev Dis Primers 2020;6:38; Curr Cardiol Rep 2021;23:98)
• ICIs can cause inflammation of any tissue (lungs, liver, colon, joints, skin, etc.)
• Common immune-related adverse events (IRAEs) above; ↑ incidence w/ combinations.
• Rare: myocarditis (can be fulminant), myositis, myelitis, uveitis, diabetes
• Workup: CT chest for dyspnea; colonoscopy and infectious workup for colitis; TSH, FT4, a.m. cortisol, glucose. Trend comprehensive metabolic panel and TSH while on ICI.
• IRAEs graded 1 (mild) to 4 (severe) (NCCN Guideline v4.2021)
ICI toxicity treatment (multidisciplinary care)
• Most IRAEs reversible with steroids
• Mild symptoms (grade 1): supportive care, can often continue Rx; moderate symptoms (grade 2): hold ICI, consider steroids; severe symptoms (grades 3–4): often requires admission to hospital, hold ICI, IV steroids, targeted therapies (eg, enteracept)
• Endocrinopathies (hypophysitis, hypothyroid) are not reversible, Rx hormone replacement
• Managing IRAEs with steroids likely does not reduce ICI efficacy (J Clin Oncol 2019;37:1927)
• If ICI is restarted, Pts at increased risk for recurrent IRAE
CHIMERIC ANTIGEN RECEPTOR (CAR)-T CELLS
Overview (Nat Rev Cancer 2021;21:145)
• Autologous T cells w/ chimeric receptor: antibody variable region fused to T cell co-stimulatory intracellular signaling domains → MHC independent Ag recognition
• CAR-T cells targeting CD19 used for ALL, DLBCL, FL, MCL. Targeting B-cell maturation antigen (BCMA) for MM. Strategies for solid tumors in development.
Toxicities
• Cytokine release syndrome (CRS): fever, HoTN, shock secondary to overwhelming cytokine release from proliferating T cells
• Immune effector cell-associated neurotoxicitiy synd. (ICANS): cerebral edema → HA, aphasia, delirium, lethargy/obtundation. Graded using ICE score (vide infra).
CAR-T Toxicity (Biol Blood Marrow Transplant 2019;25:625) |
|||
Type |
Grade |
Mechanism & Manifestations |
Treatment |
CRS |
1 |
Fever (≥38°C) ± other sx |
Supportive (fluids, O2), ± steroids ± anti-IL-6 (tocilizumab or siltuximab), depending on severity |
2 |
Fever + HoTN not needing pressors ± O2 by nasal cannula |
||
3 |
Fever + pressor ± O2 > nasal cannula |
||
4 |
Fever + >1 pressor ± ⊕ pressure vent |
||
ICANS |
1 |
ICE 7–9 pts |
Steroids, anticonvulsants for seizures |
2 |
ICE 3–6 pts |
||
3 |
ICE 0–2 pts |
||
4 |
Unarousable & unable to perform ICE |
||
ICE score: orientation (4 pts), name 3 objects (3 pts), follow commands (1 pt), write sentence (1 pt), count backward from 100 by 10 (1 pt)
ONCOLOGIC EMERGENCIES
FEVER AND NEUTROPENIA (FN) (NCCN Guidelines v.1.2021)
Definition
• Fever: single oral temp ≥38.3°C (101°F) or ≥38°C (100.4°F) for ≥1 h
• Neutropenia: ANC <500 cells/µL or <1000 cells/µL with predicted nadir <500 cells/µL
Pathophysiology and microbiology
• Predisposing factors: catheters, skin breakdown, GI mucositis, obstruction (lymphatics, biliary tract, GI, urinary tract), immune defect associated with malignancy
• Often thought due to seeding of bloodstream by GI flora, eg, GNRs (esp. P. aeruginosa)
• Neutropenic enterocolitis (typhlitis): RLQ pain, watery/bloody diarrhea, cecal wall thickening
• Gram ⊕ infections have recently become more common (60–70% of identified organisms)
• Fungal superinfection often results from prolonged neutropenia & antibiotic use
Prevention (only if intermediate or high-risk)
• Bacterial: consider FQ if neutropenic esp steroids+ALL; ↓ mortality (Cochrane 2012 CD004386)
• Fungal: consider during neutropenia in blood cancers (posa/fluconazole, micafungin)
• Viral: consider during active Rx in blood cancers (acyclovir, famciclovir, valacyclovir)
Role of hematopoietic growth factors (NCCN Guidelines v.4.2021)
• Granulocyte (G-CSF) and granulocyte-macrophage (GM-CSF) colony-stimulating factors can be used (but not in AML) as 1° Ppx when expected FN incidence >20% or as 2° Ppx after FN has occurred in a prior cycle (to maintain dose-intensity for curable tumors)
• CSFs ↓ rate of FN but have not been shown to affect mortality (Cochrane 2014 CD003039)
Diagnostic evaluation
• Exam: skin, oropharynx, lung, perirectal area, surgical & catheter sites; avoid rectal exam
• Studies: U/A, blood (periph & through each indwelling catheter port), urine, & sputum cx; for localizing s/s → ✓ stool (C. diff, cx), peritoneal fluid, CSF (rare source)
• Imaging: CXR; for localizing s/s → CNS, sinus, chest or abdomen/pelvis CT
• Caveats: neutropenia → impaired inflammatory response → exam and radiographic findings may be subtle; absence of neutrophils by Gram stain does not r/o infection
Risk stratification (factors that predict lower risk)
• History: outPt, ECOG 0–1, age <60 y, solid tumor, no sx, no major comorbidities, no h/o fungal infection, MASCC Risk Index ≥21 (Support Care Cancer 2013;21:1487)
• Exam: temp <39 °C, no tachypnea, no HoTN, no Δ MS, no dehydration
• Labs: ANC >100 cells/µL, anticipated duration of neutropenia ≤100 cells/µL <7 d
Initial antibiotic therapy (NCCN Guidelines v.1.2021)
• Empiric regimens should include antipseudomonal activity; consider VRE coverage if ⊕
• Low risk: PO abx or home IV abx may be considered in select Pts (JCO 2013;31:1149)
PO options: cipro+amoxicillin-clavulanate; levofloxacin; moxifloxacin (avoid if FQ Ppx)
• High risk: hospital admission & IV abx; monotherapy preferred
options: cefepime, imipenem, meropenem, piperacillin/tazobactam, ceftazidime
• Antifungal Rx added for neutropenic fever ≥4 d despite abx: micafungin, liposomal amphotericin B, caspofungin, anidulafungin, voriconazole, & posaconazole are options
Modification to initial antibiotic regimen based on site-specific evaluation
• Vancomycin only if HoTN, PNA, clinically apparent catheter-related or soft-tissue infxn, gram ⊕ BCx, mucositis + on quinolone Ppx; d/c when BCx ⊖ × 48 h for GPCs
• Mouth/esoph (ulcer, thrush): anaerobic (if necrotizing), anti-HSV and/or antifungal Rx
• Sinus/nasal: add vanc if periorbital cellulitis, ampho if concern for Aspergillus/Mucor
• Abd pain/diarrhea: PO vanc if concern for C. diff; ensure adequate anaerobic coverage
• Lung infiltrates: consider atypical coverage; vanc/linezolid if c/f MRSA; TMP/SMX if c/f PCP
• CNS: ID consult; empiric meningitis Rx (incl. Listeria), high-dose acyclovir for encephalitis
Duration of therapy
• Known source: complete standard course (eg, 14 d for bacteremia)
• Unknown source: continue antibiotics until afebrile and ANC >500 cells/µL
• Less clear when to d/c abx when Pt is afebrile but prolonged neutropenia
SPINAL CORD COMPRESSION
Clinical manifestations (Lancet Neuro 2008;7:459)
• Metastases located in vertebral body extend and cause epidural spinal cord compression
• Prostate, breast, and lung cancers are most common, followed by RCC, NHL, myeloma
• Site of involvement: thoracic (60%), lumbar (25%), cervical (15%)
• Signs and symptoms: pain (>95%, precedes neuro Δs), weakness, autonomic dysfunction (urinary retention, ↓ anal sphincter tone), sensory loss
• Always take back pain in Pts w/ cancer seriously. Urgent whole-spine MRI; CT if unable.
• Do not wait for neurologic signs to develop before initiating evaluation b/c duration & severity of neuro dysfunction before treatment are best predictors of neurologic outcome
Treatment (NEJM 2017;376:1358)
• Dexamethasone (10 mg IV × 1 STAT, then 4 mg IV or PO q6h)
initiate immediately while awaiting imaging if back pain + neurologic deficits
• Emergent RT or surgical decompression if confirmed compression/neuro deficits
• Surgery + RT superior to RT alone for neuro recovery in solid tumors (Lancet 2005;366:643)
• If pathologic fracture causing compression → surgery; if not surgical candidate → RT
TUMOR LYSIS SYNDROME
Clinical manifestations (NEJM 2011;364:1844)
• Large tumor burden or a rapidly proliferating tumor → spontaneous or chemotherapy-induced release of intracellular electrolytes and nucleic acids
• Most common w/ treatment of high-grade lymphomas (Burkitt’s) and leukemias (ALL, AML, CML in blast crisis). Very rare w/ solid tumors; rarely due to spont. necrosis.
• Electrolyte abnormalities: ↑ K, ↑ uric acid, ↑ PO4 → ↓ Ca; renal failure (urate nephropathy)
Prophylaxis
• Allopurinol 300 mg qd to bid PO or 200–400 mg/m2 IV (adjusted for renal fxn) & aggressive hydration prior to beginning chemotherapy or RT
Treatment
• Avoid IV contrast and NSAIDs; treat hyperK, hyperPO4, and only symptomatic hypoCa
• Allopurinol + aggressive IV hydration ± diuretics to ↑ UOP for goal 80–100 cc/h
• Rasburicase (0.1–0.2 mg/kg or 6 mg IV fixed dose) for ↑↑ uric acid esp. in aggressive malig (JCO 2003;21:4402; Acta Haem 2006;115:35). Avoid in G6PD def (hemolytic anemia). Consider G6PD testing in Jehovah’s Witnesses especially if Black (12% prevalence).
• Hemodialysis may be necessary; early renal consultation for renal insufficiency or ARF
CHEMO SIDE EFFECTS
Nausea & vomiting common (NEJM 2016;374:1356; 375:134 & 177)
Select Adverse Effects from Chemotherapy* |
||
Toxicity |
Common Agents |
Comments |
Cardiotoxicity (NEJM 2016;375;1457) |
Anthracyclines |
Dose-dependent CMP; ✓ EF pre-Rx |
5-FU |
Spasm → ischemia; CCB may prevent |
|
Trastuzumab |
CMP, esp w/ anthracycline, TTE’s for EF |
|
Tyrosine kinase inhib (TKI) |
QTc prolongation, CMP, angina |
|
Cyclophosphamide |
Myopericarditis (esp. in BMT) |
|
Cisplatin |
AKI → HypoMg / HyperK → arrhythmia |
|
Pulmonary (Sem Oncol 2006;33:98) |
Busulfan |
~8% fibrosis or DAH; if severe → steroids |
Bleomycin |
~10% IPF; ✓ PFTs pre-Rx; Rx: steroids |
|
TKI (esp. dasatinib) |
Pleural effusion |
|
Cyclophosphamide |
Pneumonitis, progressive fibrosis; Rx: d/c |
|
Bevacizumab |
Pulm. hemorrhage (esp. lung SCC) |
|
Nephrotoxicity/ urologic toxicity |
Platinum Rx (cisplatin) |
Esp. proximal tubule; pretreat w/ IV saline |
Methotrexate |
Via deposition; Rx: alkalinize urine, IVF |
|
Cyclophosphamide |
Hemorrhagic cystitis; Rx: Mesna |
|
Neurotoxicity (Sem Oncol 2006;33:324) |
Platinum Rx (cisplatin) |
“Stocking-glove” neuropathy; ototoxicity |
Cytarabine |
Cerebellar toxicity (irreversible 5–10%) |
|
Methotrexate (esp. intrathecal) |
Late leukoenceph, meningitis; reverse w/ intrathecal glucarpidase, leucovorin |
|
Ifosfamide |
Enceph; Rx: methylene blue, thiamine |
|
Taxanes, vincristine |
Sensorimotor long fiber neuropathy |
|
Hepatotoxicity (Sem Oncol 2006;33:50) |
TKI (eg, imatinib, nilotinib) |
↑ LFTs, rarely necrosis; Rx: d/c ± steroids |
Gemcitabine |
Common ↑ ALT/AST; ↓ dose if ↑ bili |
|
Methotrexate |
↑ ALT/AST, rarely fibrosis |
|
Dermatologic |
TKI (eg, imatinib) |
Dermatitis, can be severe (eg, SJS) |
*See “ImmunoRx” section for cytokine release and immune effector cell-associated neurotoxicity syndromes
Table of contents
- Cover
- Title Page
- Copyright Page
- Contents
- Contributing Authors
- Foreword
- Preface
-
CARDIOLOGY
- Electrocardiography
- Chest Pain
- Noninvasive Evaluation of CAD
- Coronary Angiography & PCI
- Stable Ischemic Heart Disease
- Acute Coronary Syndromes
- PA Catheter and Tailored Therapy
- Heart Failure
- Cardiomyopathies
- Valvular Heart Disease
- Pericardial Disease
- Hypertension
- Aortic Aneurysms
- Acute Aortic Syndromes
- Arrhythmias
- Atrial Fibrillation
- Syncope
- Cardiac Rhythm Management Devices
- Cardiac Risk Assessment for Noncardiac Surgery
- Peripheral Artery Disease
-
PULMONARY
- Dyspnea
- Pulmonary Function Tests
- Asthma
- Anaphylaxis
- Chronic Obstructive Pulmonary Disease
- Solitary Pulmonary Nodule
- Hemoptysis
- Bronchiectasis
- Cystic Fibrosis
- Interstitial Lung Disease
- Pleural Effusion
- Venous Thromboembolism
- Pulmonary Hypertension
- Respiratory Failure
- Mechanical Ventilation
- Acute Respiratory Distress Syndrome
- Sepsis and Shock
- Toxicology
- Lung Transplant
- GASTROENTEROLOGY
- NEPHROLOGY
-
HEMATOLOGY-ONCOLOGY
- Anemia
- Disorders of Hemostasis
- Platelet Disorders
- Coagulopathies
- Hypercoagulable States
- Disorders of Leukocytes
- Transfusion Therapy
- Myelodysplastic Syndromes
- Myeloproliferative Neoplasms
- Leukemia
- Lymphoma and CLL
- Plasma Cell Dyscrasias
- Hematopoietic Stem Cell Transplantation
- Lung Cancer
- Breast Cancer
- Prostate Cancer
- Colorectal Cancer
- Pancreatic Tumors
- Other Solid Tumors
- Immunotherapy & Cellular Therapy
- Oncologic Emergencies
- Chemo Side Effects
- INFECTIOUS DISEASES
- ENDOCRINOLOGY
-
RHEUMATOLOGY
- Approach to Rheumatic Disease
- Rheumatoid Arthritis
- Adult-Onset Still’s Disease & Relapsing Polychondritis
- Crystal Deposition Arthritides
- Seronegative Spondyloarthritis
- Infectious Arthritis & Bursitis
- Connective Tissue Diseases
- Systemic Lupus Erythematosus
- IgG4-Related Disease
- Vasculitis
- Autoinflammatory Syndromes
- Amyloidosis
- NEUROLOGY
- CONSULTS
- APPENDIX
- ABBREVIATIONS
- INDEX
- PHOTO INSERTS
- ACLS